 Resumen
Aquí se revisan algunos aspectos nuevos sobre el síndrome metabólico. La primera parte de este artículo analiza por qué la epidemia de síndrome metabólico, diabetes mellitus y enfermedad cardiovascular afecta en forma creciente a la población de países en vías de desarrollo. Específicamente se revisan las hipótesis que plantean la relación entre la resistencia a la insulina generada en el útero y la desnutrición intrauterina por desnutrición materno-fetal o disfunción placentaria. Ambas condiciones prenatales son problemas médico-económicos, muy frecuentes en poblaciones pauperizadas, que se traducen en altos índices de bajo peso al nacer. En la segunda parte de esta revisión se presentan nuevas pruebas epidemiológicas que sustentan la validez del síndrome metabólico como herramienta clínica diagnóstica y de pronóstico. Se hace especial énfasis en el artículo de Canoy y col., extraída del estudio epidemiológico de Norfolk. En esa publicación, los autores demuestran, en un diseño prospectivo con seguimiento de 9.1 años en más de 20 000 individuos, que el índice cintura abdominal/cadera, es el mejor marcador clínico de adiposidad corporal para la predicción de un evento cardiovascular. Este hallazgo epidemiológico tiene un gran valor clínico. Finalmente, en la tercera parte de esta puesta al día, se resumen los datos clave de investigación básica, sobre los mecanismos proinflamatorios de la obesidad abdominal (intercomunicación adipocito disfuncional-monocito). En forma complementaria se presenta información de reciente aparición, con foco en la adiponectina. Los mecanismos intrínsecos que hacen a la adiponectina una molécula cardioprotectora y cerebroprotectora son analizados en esta última sección.
Palabras clave
síndrome metabólico, resistencia a la insulina, hemoglobina glucosilada fetal, adipocito disfuncional, adiponectina
Resumen
Aquí se revisan algunos aspectos nuevos sobre el síndrome metabólico. La primera parte de este artículo analiza por qué la epidemia de síndrome metabólico, diabetes mellitus y enfermedad cardiovascular afecta en forma creciente a la población de países en vías de desarrollo. Específicamente se revisan las hipótesis que plantean la relación entre la resistencia a la insulina generada en el útero y la desnutrición intrauterina por desnutrición materno-fetal o disfunción placentaria. Ambas condiciones prenatales son problemas médico-económicos, muy frecuentes en poblaciones pauperizadas, que se traducen en altos índices de bajo peso al nacer. En la segunda parte de esta revisión se presentan nuevas pruebas epidemiológicas que sustentan la validez del síndrome metabólico como herramienta clínica diagnóstica y de pronóstico. Se hace especial énfasis en el artículo de Canoy y col., extraída del estudio epidemiológico de Norfolk. En esa publicación, los autores demuestran, en un diseño prospectivo con seguimiento de 9.1 años en más de 20 000 individuos, que el índice cintura abdominal/cadera, es el mejor marcador clínico de adiposidad corporal para la predicción de un evento cardiovascular. Este hallazgo epidemiológico tiene un gran valor clínico. Finalmente, en la tercera parte de esta puesta al día, se resumen los datos clave de investigación básica, sobre los mecanismos proinflamatorios de la obesidad abdominal (intercomunicación adipocito disfuncional-monocito). En forma complementaria se presenta información de reciente aparición, con foco en la adiponectina. Los mecanismos intrínsecos que hacen a la adiponectina una molécula cardioprotectora y cerebroprotectora son analizados en esta última sección.
Palabras clave
síndrome metabólico, resistencia a la insulina, hemoglobina glucosilada fetal, adipocito disfuncional, adiponectina
Abstract
Some new insights on the metabolic syndrome are reviewed. The reasons why epidemic metabolic syndrome, diabetes mellitus, and cardiovascular disease are increasingly affecting poor economies are discussed in the first part of this article. Hypothesis on the specific relation between in-utero insulin resistance and in-utero undernutrition due to mother-fetal undernutrition and/or placental dysfunction are reviewed. Both prenatal conditions, which are medical and economic problems, are often encountered in pauperized populations and turn into elevated rates of low birth weight. In the second part of this revision, new epidemiological evidence on the value of metabolic syndrome as a tool for diagnosis and prognosis is presented. Special emphasis has been placed on the MPhil publication, based on Norfolk's epidemiological study. In this publication the authors have demonstrated, using a prospective design in 20 000 subjects who were followed for 9.1 years, that waist/hip ratio is the best clinical marker of body adiposity in order to predict cardiovascular events. This finding is of great clinical value. Finally, in the third part of this update, key data from basic research on pro-inflammatory mechanisms in abdominal obesity (dysfunctional adipocyte-monocyte intercommunication) are summarized. Also, new information focused on adiponectin is presented. The mechanisms intrinsic to adiponectin, which make this molecule protective for heart and brain, are discussed in this last section.
Key words
metabolic syndrome, insulin resistance, fetal glycosylated hemoglobin, dysfunctional adipocytes, adiponectin
Artículo completo
SINDROME METABOLICO, EL CONTINUUM CARDIOLETAL
(especial para SIIC © Derechos reservados)
Desnutrición intrauterina y obesidad infantil, la paradoja del síndrome metabólico
Resulta paradójico que la epidemia de obesidad, diabetes mellitus (DBT) y enfermedad cardiovascular (ECV) afecte principalmente a la población de países con economías en desarrollo. Sin embargo, dicho fenómeno tiene al menos dos puntos de origen. El primero es la alta prevalencia de desnutrición prenatal, y el segundo es la modificación inapropiada del estilo de vida de las poblaciones de economías pauperizadas.
Hattersley y Tooke1 propusieron que la resistencia a la insulina (RI) es una alteración genética primaria (esencial) y que por ende se manifiesta aun desde la etapa prenatal. Por el contrario, Phillips-Barker y Bagby2 plantearon la hipótesis de que la RI es una condición asociada a la desnutrición prenatal, sea por desnutrición materno-fetal, por disfunción placentaria o por ambas. En su hipótesis, estos autores afirman que, bajo circunstancias de desnutrición intrauterina, los bebés en desarrollo, por un fenómeno conocido como "plasticidad genética", crean de forma reactiva-adaptativa, un estado de resistencia periférico-muscular esquelética a la insulina. Este proceso, teleológicamente, incrementa el flujo de glucosa de la periferia hacia el cerebro. Este flujo neurocéntrico de glucosa, si bien privilegia el desarrollo neuronal, también determina hipodesarrollo musculoesquelético, que se traduce al nacimiento como bajo peso. Dicha "plasticidad genética", causante de resistencia muscular intrauterina a la insulina, es la que en etapas posnatales puede mantenerse quiescente o bien manifestarse en forma subclínica o clínica, principalmente si el individuo presenta obesidad abdominal en su infancia o en su adolescencia.
La asociación entre bajo peso al nacer y RI originada en el útero y diagnosticada al nacimiento está bien documentada. Pfab y col.3 publicaron en 2006 un estudio en el que presentaron los resultados de un muestreo de 1 295 determinaciones de hemoglobina (Hb) glucosilada total en sangre fetal y materna, tomadas durante el nacimiento. Estos autores informaron que el nivel de Hb glucosilada total fetal y el cociente Hb glucosilada total fetal/Hb glucosilada total materna, guardan una relación inversamente proporcional al peso del bebé al nacimiento. Después del análisis multivariado, esta relación fue estadísticamente muy significativa, con un valor de p < 0.0001. Es decir que a mayor concentración de Hb glucosilada total fetal, menor peso al nacer. Estas observaciones documentan fehacientemente la relación entre el bajo peso al nacer y la RI, traducida como incremento prenatal de Hb glucosilada total en el recién nacido.
Así entonces, un bebé cuyo desarrollo intrauterino fue desfavorable por desnutrición o disfunción placentaria, tendrá una alta probabilidad de nacer con bajo peso y con un estado subyacente de resistencia musculoesquelética a la insulina.
En qué grado el fenómeno referido se liga a la epidemia de obesidad visceral, síndrome metabólico (SM), DBT y ECV en edades cada vez más tempranas es difícil de establecer con precisión, sin embargo, de acuerdo con la visión de algunos expertos en el tema, es muy lógico que así sea.
Ludwing,4 a propósito de dos artículos recientes sobre la obesidad en la niñez y en la adolescencia, y el riesgo de enfermedad cardiovascular en la adultez temprana,5,6 escribió un análisis en el que propone que dicho fenómeno inició en la década de 1970 con una "primera ola" (Figura 1), identificada por un cambio del patrón de estilo de vida infantil, caracterizado por incremento en la carga calórica de la alimentación y reducción en el gasto energético. Este cambio de estilo de vida infantil ocasionó un significativo incremento en los promedios de peso corporal en ambos sexos y en todos los estratos socioeconómicos, si bien con una tendencia a ser más notable en ciertas comunidades menos favorecidas.

Esa generación de "niños gordos", dio pie a la actual "segunda ola", perfilada por la incidencia creciente de obesidad visceral en niños, adolescentes y adultos jóvenes, causal –por los mecanismos que se analizarán más adelante– de SM, hígado graso, DBT, y otras consecuencias físicas y psicológicas que conlleva el incremento de la adiposidad corporal.
La "tercera ola" se predice que tendrá lugar entre 2025 y 2035 y se identificará por una reducción en la expectativa de vida. Así, los adolescentes y los adultos jóvenes de hoy sufrirán invalidez o morbimortalidad cardiovascular tempranas; la de ellos será la primera generación de la raza humana que sobrevivirá menos que su antecesora.
Finalmente, y en forma hipotética, la "cuarta ola" podría presentarse en caso de que nuestro medio ambiente obesogénico no sea abatido. En esas circunstancias se anticipa que por un fenómeno similar al de la "plasticidad genética", el organismo humano transformará en forma reactiva-adaptativa su estructura genética. De esa forma, las siguientes, serán generaciones de humanos obesos "adaptados" a su entorno, a costa de un promedio de vida de 50-60 años (comparado con el actual, de 75 años). Esta hipótesis es denominada de la "obesidad transgeneracional" y definitivamente es alarmante.
Nuevas pruebas epidemiológicas del papel nocivo de la obesidad abdominal
Dekker7 y Wilson8 publicaron en 2005 dos de los artículos epidemiológicos más importantes sobre la relación entre SM y ECV y DBT. Ambos autores demostraron que tanto en la población americana como europea, la existencia de variables diagnósticas de SM, correlaciona en forma lineal con la magnitud del riesgo de evolucionar hacia DBT, ECV o ambas. El incremento del riesgo requiere sólo de la presencia de una variable, y crece en forma directamente proporcional al número de variables acumuladas, llegando a ser tan alto como de 5.95 en comparación al riesgo basal, para un evento coronario a 8 años, en mujeres con tres o más criterios de SM8 (Figura 2).
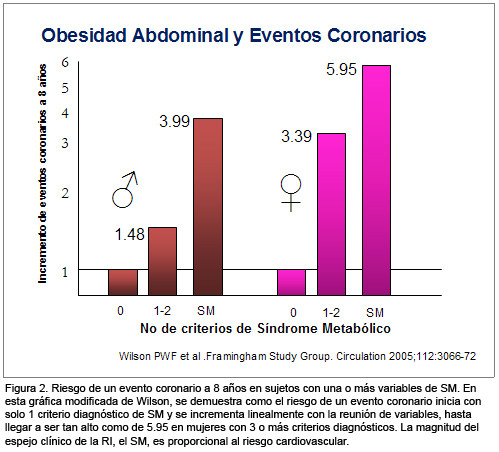
Nuevas publicaciones sustentan a aquellas que en el primer quinquenio de nuestro siglo apuntalaron el valor clínico del concepto de SM.
De ellas, una de las más importantes por su diseño prospectivo y su esencia clínica, es la publicada en diciembre de 2007 por Canoy y col.9 Ellos analizaron en forma prospectiva la relación de tres diferentes marcadores clínicos de adiposidad corporal (índice de masa corporal, circunferencia de cintura abdominal e índice cintura/cadera), con la incidencia de enfermedad cardíaca coronaria. Su cohorte de estudio fueron 24 508 hombres y mujeres de entre 45 y 79 años al momento de la inclusión y el seguimiento promedio fue de 9.1 años. El resultado más contundente fue el siguiente.
El mejor marcador pronóstico (valor predictivo positivo, independiente de otras variables y consistente en diferentes subpoblaciones) de un evento cardíaco coronario fue la relación cintura/cadera, el riesgo de un evento cardíaco coronario fue de 1.55 en hombres y de 1.91 en mujeres, ubicados en los quintilos más altos, comparados con aquellos localizados en los quintilos más bajos para cada sexo. El índice cintura/cadera también mostró una asociación lineal, independiente y significativa con otros factores de riesgo cardiovascular, especialmente edad, presión arterial sistólica, colesterol total y hábito de fumar. Finalmente, la predicción de un evento cardíaco coronario fue más sólida en la subpoblación que, al momento de su inclusión en la cohorte no tenía antecedente de ECV (Figura 3).
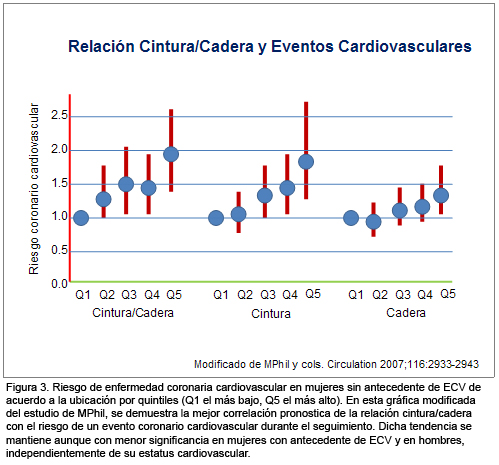
El valor pronóstico de la circunferencia de la cintura abdominal se encontró en un punto intermedio y el índice de masa corporal fue el marcador de menor valor pronóstico.
Los resultados referidos guardan una estrecha relación que concuerda con los ampliamente conocidos del estudio INTERHEART.10 Ambos estudios son una demostración epidemiológica del papel nocivo de la grasa abdominal versus el papel protector de la grasa subcutánea medida en la cadera.
En forma sencilla y práctica se puede establecer un concepto clínico muy valioso. A mayor perímetro abdominal (grasa mala), mayor riesgo de un evento cardiovascular y a mayor perímetro de cadera (grasa buena), menor riesgo de un evento cardiovascular, siendo la relación perímetro abdominal/perímetro de cadera > 0.9, el valor a partir del cual el riesgo cardiovascular se incrementa.
Nuevos mecanismos patogénicos de la obesidad abdominal
La validez y la utilidad del concepto de SM, ha sido analizada ampliamente.11 Taskinen, en su reciente editorial de ATVB, "¿Es el síndrome metabólico la principal amenaza para la salud humana en el siglo XXI?",12 engloba en la siguiente frase la esencia actual de su análisis: "El SM es un conjunto de factores de riesgo cardiometabólico que coexisten en individuos con obesidad central y resistencia a la insulina. Aunque ha existido controversia en relación con los criterios y el concepto de SM, las definiciones actuales del NCEP-ATP-III y de la IDF brindan herramientas útiles para la identificación de individuos con un riesgo cardiometabólico elevado".
Así, la aglutinación mayor a la esperada por el azar de: obesidad abdominal, condición sine qua non para el diagnóstico de SM; niveles de glucosa mayores de 100 mg/dl; colesterol asociado a lipoproteínas de alta densidad (HDLc) por debajo de 40 mg o 50 mg/dl, en hombres y mujeres, respectivamente; niveles de triglicéridos por arriba de 150 mg/dl; tensión arterial mayor de 130-85 mm Hg (Figura 4), es un reflejo clínico de fácil detección, el cual traduce un estado de RI, causado por la obesidad abdominal y causante de desregulación glucémica, desregulación lipídica y desregulación hemodinámica. Estas alteraciones en la fisiología del metabolismo de la glucosa, los lípidos y la tensión arterial son a su vez la traducción de la resistencia a la insulina posreceptor en las células del eje metabólico, hepatocito/miocito/adipocito, y en la célula endotelial.11
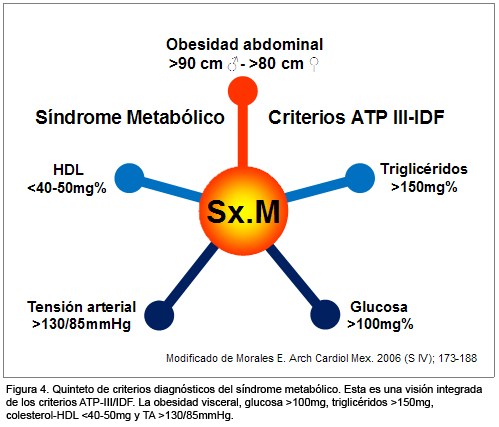
Como se analizó en el apartado previo, si bien la esencia de la resistencia a la insulina puede ser genética primaria,1 genética reactiva o adaptativa,2 o sólo adquirida, es un hecho que su disparador más común es el proceso de lipotoxicidad e inflamación, iniciado por la disfunción de los adipocitos "obesos" y su nociva intercomunicación con los monocitos del estroma visceral abdominal.11,13 Proceso éste retroalimentado, sostenido y amplificado por los cada vez mejor conocidos mecanismos proinflamatorios que se analizan en el siguiente apartado (Figura 4).
Con una visión clínica, la virtud del concepto SM es brindar al médico en su consultorio una herramienta de fácil manejo que le permita detectar individuos que hace un decenio se consideraban fuera de riesgo ("obesidad = problema estético") y de los cuales ahora sabemos que a medida que acumulan variables de SM, su riesgo de evolucionar hacia DBT, ECV o ambos se incrementa linealmente. Es pues una observación contundente el que la suma de obesidad abdominal más cualquier otra prueba de RI (SM), eleva proporcionalmente el riesgo de algún evento cardiovascular o de muerte. No es necesario reunir las tres variables diagnósticas, el riesgo inicia con una variable y crece proporcionalmente.7-9
De lo anterior se desprende el razonamiento lógico que liga de manera causal la obesidad abdominal con la RI y la DBT y la ECV.
A continuación desglosaremos datos recientes sobre la fisiopatología y la anatomopatología de la obesidad abdominal como causa de RI, DBT y ECV aterotrombótica.
Adipocito-monocito, el binomio "culpable"
En condiciones fisiológicas el adipocito es una célula con la estructura transcripcional genética y enzimática requeridas para sintetizar triglicéridos a partir de ácidos grasos y glicerol. La capacidad del adipocito para sintetizar triglicéridos e impedir su hidrólisis está determinada fundamentalmente por la síntesis, la expresión y la función de los receptores de membrana para insulina. De esta forma un adipocito maduro es una célula sensible a la insulina, cuyos iconos fisiológicos son la lipogénesis, la antilipólisis y la producción de adiponectina (véase más adelante).
Gustafson, en una revisión reciente,14 presenta en forma amplia pero precisa el conocimiento más depurado sobre el "tejido adiposo inflamado", como causa subyacente de SM y aterosclerosis. Aquí los aspectos más importantes de esta revisión (Figura 5).
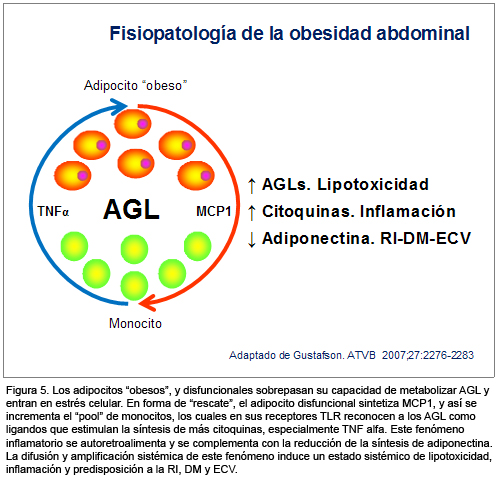
Evolutivamente, el adipocito y el macrófago son dos estirpes celulares con un origen común. La evolución del preadipocito hacia adipocito maduro requiere la represión de sus factores de transcripción genética primitivos "proinflamatorios símil macrófago", y la activación de sus factores de transcripción genética (PPAR-γ, entre otros) "lipogénicos, antilipolíticos e inductores de la síntesis de adiponectina".
A su vez, los monocitos pueden desdiferenciarse hacia células con función "símil adipocito".
El proceso de desdiferenciación del adipocito implica que el adipocito maduro reprima sus factores de transcripción genética de célula madura y reactive los de célula indiferenciada o primitiva (preadipocito), con comportamiento "proinflamatorio símil macrófago". Por otra parte, en el estroma adiposo visceral existen células mesenquimales que pueden diferenciarse hacia adipocitos maduros o bien interrumpir su diferenciación (indiferenciación) y "estacionarse" en la etapa de preadipocito.
Pues bien, la obesidad abdominal, por mecanismos que se describen más adelante, es el factor causal más frecuente de indiferenciación y desdiferenciación adipocitaria.
El exceso de adiposidad visceral abdominal favorece la disfunción cualitativa y cuantitativa del adipocito, contenido en el estroma adiposo visceral. Esta disfunción adipocitaria se inicia con la incapacidad del adipocito para metabolizar el exceso de ácidos grasos incorporados en su interior, fenómeno que le provoca un estado de "estrés celular". El estrés celular del adipocito, especialmente el del retículo sarcoplásmico, estimula la desinhibición de factores de transcripción genética proinflamatoria, como el factor nuclear kappa B (NFκB). En el adipocito disfuncional, este factor de transcripción genética desinhibido induce, entre otras, la síntesis de la citoquina MCP1 (monocyte chemoatractant protein 1).
Este mecanismo, reactivo al estrés celular, tiene el objetivo de atraer por diapédesis a monocitos hacia el estroma adiposo visceral, en un intento de que éstos, al adoptar sus características "símil adipocito", incorporen por fagocitosis el exceso de ácidos grasos libres que el adipocito disfuncional no ha sido capaz de metabolizar. Como todo mecanismo de compensación fisiológica, tiene un límite.
Con el correr del tiempo, la progresión de la obesidad visceral abdominal "satura" la capacidad fisiológica del adipocito y del monocito-macrófago para contener intracelularmente a los ácidos grasos libres y así inicia una segunda fase de retroalimentación proinflamatoria.
Los ácidos grasos libres y en exceso, especialmente los saturados, por vía paracrina, son agonistas de los receptores tipo toll (RTT) de los monocitos del estroma adiposo visceral. La principal molécula producida por el monocito ante dicha estimulación, es el facto de necrosis tumoral alfa (FNT-α).
El FNT-α, igualmente por vía paracrina, es agonista de los receptores específicos para esta citoquina, entonces ya expresados en el adipocito indiferenciado o desdiferenciado (FNT-αr1 y FNT-αr2). Este estímulo induce los siguientes cambios en la expresión fenotípica del adipocito:
a) incrementa la insensibilidad a la insulina del adipocito maduro al "quemar" por transactivación los sustratos de la cascada enzimática dependiente de fosfatidil inositol 3 quinasa (PI3k);
b) frena la diferenciación de preadipocitos hacia adipocitos al reprimir la expresión de factores de transcripción como el PPAR-γ;
c) activa en los preadipocitos y en los adipocitos desdiferenciados la cascada enzimática dependiente de la fosfoquinasa activada por mitógenos (MAPk), cuyo fin último es desinhibir al factor de transcripción NFκB. Esta molécula, al desligarse de su inhibidor (IκK), migra hacia el núcleo celular y orquesta la transcripción de más de cien moléculas proinflamatorias.
En un inicio, este proceso proinflamatorio está limitado al estroma adiposo visceral y se caracteriza por insensibilidad del adipocito visceral a la insulina, liberación incrementada de ácidos grasos hacia el estroma visceral, producción de numerosas moléculas proinflamatorias e inhibición de la producción de otras antiinflamatorias, entre las que se destaca la adiponectina.
Sin embargo, la conexión circulatoria del estroma adiposo visceral con el hígado, a través de la vena porta, es el canal para que dicho ambiente proinflamatorio visceral-abdominal sea amplificado y difundido sistémicamente, traduciéndose en un estado crónico de inflamación subclínica sistémica, con múltiples consecuencias, y que a manera de resumen pueden enunciarse de la siguiente forma:11
a) Resistencia endotelial a la insulina con disfunción endotelial. Activación proinflamatoria celular endotelial con atracción, adhesión y agregación leucoplaquetaria al endotelio; permeabilidad y activación monocitaria en el subendotelio; vasoconstricción por activación celular muscular lisa y disfunción de células endoteliales progenitoras.
b) Resistencia hepática y muscular a la insulina con desregulación lipídica y glucémica. Lipólisis sistémica; dislipidemia mixta aterogénica; estrés oxidativo por acumulación de productos intermedios de glucosilación.
c) Hiperinsulinemia y disfunción cualitativa y cuantitativa de la célula beta. Mayor hiperglucemia; retención de Na+ y agua, hiperactivación de los SNS-SRAA y retroalimentación paradójica de cascadas proinflamatoria determinadas por el estado de RI e hiperinsulinemia "filantrópica".
d) Diabetes mellitus. Resistencia sistémica a la insulina, lesión cualitativa y cuantitativa de células beta pancreáticas.
d) Aterotrombosis. Aterogénesis multifactorial, neovascularización subintimal, ruptura de placa ateromatosa, trombosis intraluminal.
Sin lugar a dudas, el papel nocivo de los ácidos grasos libres y de las diferentes citoquinas proinflamatorias originadas en el tejido adiposo abdominal disfuncional, y difundidas a toda la economía vía portal-hepática, ha sido, durante los últimos cinco años, el aspecto más estudiado en la fisiopatología de la RI y sus consecuencias.
A continuación se revisan algunos datos recientes sobre la importancia de la otra cara de la RI, es decir la regulación negativa en la síntesis y expresión de adipocitoquinas "buenas", muy especialmente de la adiponectina.
Adiponectina. Una nueva opción diagnóstico-terapéutica
Es interesante cómo nuestro conocimiento se complementa ahora con publicaciones sobre el papel de la adiponectina. Partiendo del hecho de que la hipoadiponectinemia es un marcador de mal pronóstico cardiometabólico,15 a continuación se presentan hallazgos recientes sobre los mecanismos que explican por qué la adiponectina es una molécula cardioprotectora y neuroprotectora. Este fenómeno es de tal magnitud, que se considera a dicha adipohormona como una muy sólida candidata para incorporarse al arsenal terapéutico de las enfermedades coronaria y cerebrovascular.
Esta visión nos permite entender ahora lo que una época reciente se consideró como la paradoja de "la carencia de una adipohormona en un ambiente de adiposidad visceral".16 La paradoja se resuelve al entender que la fisiopatología de la RI causada por la obesidad abdominal se caracteriza tanto por un nocivo incremento de las adipocitoquinas proinflamatorias como por una –también nociva– reducción de la adipocitoquina antiinflamatoria por excelencia, la adiponectina.
En estudios epidemiológicos se demostró sólida y repetidamente la correlación entre hipoadiponectinemia y obesidad abdominal. Asimismo se documentó la relación entre DBT y ECV, con niveles bajos de adiponectina. Como apoyo a toda esa evidencia epidemiológica, ahora empieza a consolidarse el conocimiento de los mecanismos intrínsecos por los cuales la adiponectina es una molécula cuya concentración se correlaciona inversamente con la presencia y el pronóstico de la ECV.17
En un sentido positivo, Shinmura y col.18 determinaron el mecanismo por el cual la adiponectina tiene un efecto cardioprotector. Partiendo de la base de que la restricción calórica se asocia a un efecto cardioprotector, incluso a la prolongación de la supervivencia, y que la restricción calórica determina elevación en la concentración de adiponectina,17 estos autores demostraron que dicho efecto cardioprotector de la restricción calórica está relacionado con la elevación "reactiva" en el nivel de adiponectina. Ellos documentaron en un modelo con animales que la fracción de alto peso molecular de la adiponectina incrementa su concentración después de períodos variables de restricción calórica y estimula la cascada enzimática de la AMPk. Lo anterior, bajo condiciones de isquemia miocárdica, estimula la captación de sustratos para la glucólisis y la producción de ATP.
Este mecanismo es una sólida explicación mecanicista de la asociación entre ayuno (restricción calórica), incremento en la concentración de adiponectina, cardioprotección y quizá prolongación de la supervivencia. En otros términos, la restricción calórica crearía un fenómeno de preacondicionamiento miocárdico a la isquemia, estimulando la síntesis de adiponectina y con ello la producción de fosfatos de alta energía a través de la estimulación de la AMPk.
Nishimura y col.19 publicaron recientemente la primera prueba de que la adiponectina circulante se une a la matriz extracelular –colágeno I y III– de los vasos arteriales de territorios cerebrales isquémicos, y protege al cerebro contra el daño isquémico, a través de la estimulación por fosforilación de la sintasa de óxido nítrico endotelial (eNOS), con incremento en la producción de óxido nítrico y en la perfusión vascular nativa y colateral hacia el territorio isquémico. Previamente se demostró que la AMPk tiene como sus sustratos en la célula endotelial la Akt y la eNOS.20,21
Estas observaciones son concordantes con las de Shinmura y col. y sustentan la teoría de que la adiponectina, al concentrase en zonas de isquemia tisular por lesión endotelio-vascular y unirse a sus receptores de membrana específicos, estimula la cascada enzimática de la AMPk, y ésta, a su vez, estimula otros sustratos enzimáticos como la eNOS con producción de óxido nítrico y sus benéficas acciones subrogadas (Figura 6).
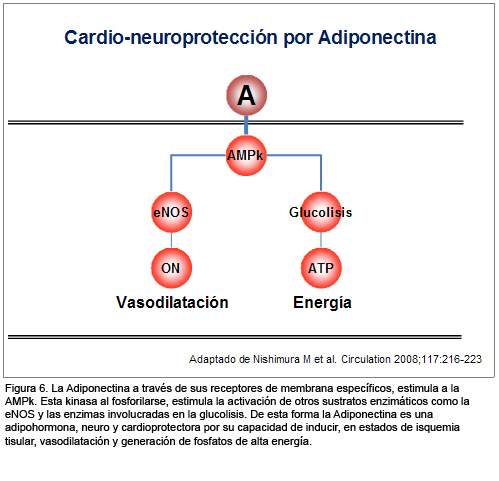
Ambos hallazgos brindan una explicación básica de la relación plenamente demostrada en estudios epidemiológicos entre hipoadiponectinemia y obesidad y sus complicaciones como DBT y ECV. Por otra parte, abren la posibilidad para estrategias terapéuticas que por medio de suplementar o estimular la síntesis de adiponectina protejan órganos como el cerebro o el corazón del daño por isquemia y reperfusión en sujetos con SM.
Conclusión
Es una hipótesis muy bien sustentada la que liga la obesidad infantil, condicionada por la alta carga calórica y la poca actividad física, como el disparador hacia la escena clínica como SM, el estado de RI de quienes durante su desarrollo prenatal desarrollaron esta condición de insensibilidad periférica a la insulina por desnutrición materno-fetal o disfunción placentaria o ambas (teoría de la plasticidad genética).
La relación entre el perímetro de la cintura abdominal/perímetro de la cadera es un sólido indicador pronóstico de ECV. Su simpleza, valor predictivo y superioridad sobre el perímetro de cintura y el IMC lo colocan como el mejor marcador clínico de adiposidad corporal.
La adiponectina, en estados de isquemia tisular, es una adipohormona con acción cardioprotectora y cerebroprotectora. La estimulación de la AMPk y de sus cascadas enzimáticas subrogadas (eNOS) es el mecanismo intrínseco por medio del cual se explica el incremento en la formación de ATP y de la vascularidad nativa y colateral dependiente de la adiponectina. De esta forma se explica la relación inversa entre los niveles de adiponectina y la incidencia y pronóstico de la ECV en sujetos con SM (a menor adiponectina, peor pronóstico).
|
Bibliografía del artículo
1. Hattersley AT, Tooke JE. The fetal insulin hypothesis: An alternative explanation of the association of low birthweigth with diabetes and vascular disease. Lancet 353:1789-1792, 1999.
2. Barker DJ, Bagby SP. Developmental antecedents of cardiovascular disease: a historical perspective. J Am Soc Nephrol 16:2537-2544, 2005.
3. Pfab T, Slowinski T, Godes M, Halle H, Priem F, Hocher B. Low birth weigth, a risk factor for cardiovascular diseases in later life, is already associated with elevated fetal glycosylated hemoglobin at birth. Circulation 114:1687-1682, 2006.
4. Ludwing DS. Childhood obesity. The shape of things to come. N Engl J Med 357:2325-2327, 2007.
5. Baker J, Olsen LW, Sorensen TIA. Childhood body mass index and the risk of coronary heart disease in adulthood. N Engl J Med 357:2329-2337, 2007.
6. Domingo KB, Coxson P, Pletcher MJ, Lightwood J, Goldman L. Adolescent overweight and future adult coronary heart disease. N Engl J Med 357:2371-2379, 2007.
7. Dekker JM, Girman C, Rhodes T, Nijpels G, Stenhouwer CDA, Bouter LM, Heine RJ. Metabolic syndrome and 10-year cardiovascular disease risk in the Hoorn study. Circulation 112:666-673, 2005.
8. Wilson PWF, DÁgostino RB, Parise H, Sullivan L, Meigs JB. Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation 112:3066-3072, 2005.
9. Canoy D, Boekholdt SM, Wareham N, Luben R, Welch A, Bingham S, Buchan I, Day N, Khaw KT. Body fat distribution and risk of coronary heart disease in men and women in the European Prospective Investigation into Cancer and Nutrition in Norfolk cohort. Circulation 116:2933-2943, 2007.
10. Yusuf S, Hawken S, Ounpuu S, Bautista L, Franzosi MG, Commerford P, Lang CC, Rumboldt Z, Onen CL, Lisheng L, Tanomsup S, Wangai P Jr, Razak F, Sharma AM, Anand SS. Obesity and the risk of myocardial infarction in 27,000 participants from 52 countries: a case-control study. Lancet 366:1640-1649, 2005.
11. Morales Villegas E. Síndrome X vs Síndrome Metabólico. Entendiendo sus coincidencias y diferencias hacia una "nueva cardiología". Arch Cardiol Mex 76 (Suppl 4):173-188, 2006.
12. Taskinen MR. Editorial. Is metabolic syndrome the main threat to human health in the twenty-first century. Atheroscler Thromb Vasc Biol 27:2275, 2007.
13. Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggarvates inflamatory changes. Role of free fatty acids and tumor necrosis factor alfa. Arterioscler Thromb Vasc Biol 25:2062-2068, 2005.
14. Gustafson B, Hammarstedt A, Anderson CX, Smith U. Inflamed adipose tissue. A culprit underlying the metabolic syndrome and atherosclerosis. Atheroscler Thromb Vasc Biol 27:2276-2283, 2007.
15. Hopkins TA, Ouchi N, Shibata R, Walsh K. Adiponectin actions in the cardiovascular system. Cardiovasc Res 74:11-18, 2007.
16. Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, HottaK, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradogical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257:79-83, 1999.
17. Dyck JRB. Editorial. The ischemic heart. Starving to stimulate the adiponectin-AMPk signaling axis. Circulation 116:2779-2781, 2007.
18. Shinmura K, Tamaki K, Saito K, Nakano Y, Tobe T, Bolli R. Cardioprotective effects of short-term caloric restriction are mediated by adiponectin via activation of AMP-activated protein kinase. Circulation 116:2809-2817, 2007.
19. Nishimura M, Izumiya Y, Higuchi A, Shibata R, Qiu J, Kudo Ch, Shin HK, Moskowitz A, Ouchi N. Adiponectin prevents cerebral-ischemic injury through endothelial nitric oxide synthase-dependent mechanisms. Circulation 117:216-223, 2008.
20. Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, Funahashi T, Walsh K. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem 279:1304-1309, 2004.
21. Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem 278:45021-45026, 2003.
Artículos publicados por el autor
(selección):
Morales Villegas EC, González García C, Hernández García HR. Obesidad. Un problema de peso. El papel terapéutico actual y maduro de la sibutramina. El peso de la evidencia. Rev Mex Cardiol. 18(18):163-172, 2007
|
|
