 Resumen
La enfermedad celíaca (EC) es un trastorno inflamatorio crónico del intestino delgado inducido por la ingestión de gluten de trigo y otras prolaminas de cereales como cebada, centeno o avena. Afecta a las personas con susceptibilidad genética, y se manifiesta por una lesión de la mucosa intestinal (con linfocitosis intraepitelial, pérdida de vellosidades y remodelación tisular), y la presencia de anticuerpos antitransglutaminasa. El modelo patogénico más aceptado se basa en la activación de una respuesta de la inmunidad adaptativa tras la estimulación de linfocitos T CD4+ mediante péptidos de gluten modificados por la enzima transglutaminasa tisular presentados junto a moléculas HLA-DQ2 o DQ8, y la producción de citoquinas y otros mediadores proinflamatorios. El gluten activa también la inmunidad innata local y mecanismos de citotoxicidad sobre el epitelio mediados por linfocitos intraepiteliales. Aunque no se conoce bien cuál es el efecto o la implicación patogénica de los anticuerpos específicos de la EC, la disponibilidad de marcadores serológicos e inmunogenéticos como herramientas diagnósticas ha propiciado el avance en el conocimiento de la EC, y la revisión de los criterios diagnósticos, especialmente en los individuos adultos con expresión mínima o atípica de la enfermedad.
Palabras clave
enfermedad celíaca, linfocitos T, péptidos de gluten, HLA-DQ2/DQ8, transglutaminasa, citoquinas, anticuerpos
Resumen
La enfermedad celíaca (EC) es un trastorno inflamatorio crónico del intestino delgado inducido por la ingestión de gluten de trigo y otras prolaminas de cereales como cebada, centeno o avena. Afecta a las personas con susceptibilidad genética, y se manifiesta por una lesión de la mucosa intestinal (con linfocitosis intraepitelial, pérdida de vellosidades y remodelación tisular), y la presencia de anticuerpos antitransglutaminasa. El modelo patogénico más aceptado se basa en la activación de una respuesta de la inmunidad adaptativa tras la estimulación de linfocitos T CD4+ mediante péptidos de gluten modificados por la enzima transglutaminasa tisular presentados junto a moléculas HLA-DQ2 o DQ8, y la producción de citoquinas y otros mediadores proinflamatorios. El gluten activa también la inmunidad innata local y mecanismos de citotoxicidad sobre el epitelio mediados por linfocitos intraepiteliales. Aunque no se conoce bien cuál es el efecto o la implicación patogénica de los anticuerpos específicos de la EC, la disponibilidad de marcadores serológicos e inmunogenéticos como herramientas diagnósticas ha propiciado el avance en el conocimiento de la EC, y la revisión de los criterios diagnósticos, especialmente en los individuos adultos con expresión mínima o atípica de la enfermedad.
Palabras clave
enfermedad celíaca, linfocitos T, péptidos de gluten, HLA-DQ2/DQ8, transglutaminasa, citoquinas, anticuerpos
Abstract
Celiac disease is a chronic inflammatory process of the small bowel induced by the ingestion of wheat gluten and other prolamines from cereals, barley, rye and oats, affecting genetically susceptible individuals, and manifested by a lesion of the intestinal mucosa (intraepithelial lymphocytosis, loss of villi, tissue remodelling), and anti-transglutaminase antibodies. The pathogenic model is based on the activation of an adaptive immune response due to the recognition of T CD4+ cells of gluten peptides modified by tissue transglutaminase and presented along with HLA-DQ2 o DQ8 molecules, production of cytokines and other inflammatory mediators. Gluten peptides also induce a response of innate immunity and epithelial cytotoxicity mediated by intraepithelial lymphocytes. Though the implication of specific antibodies in the pathogenesis of CD is still unclear, the availability of serological and immunogenetic markers as diagnostic tools has brought about advance/progress in our knowledge about DC, and the revision of diagnostic criteria, particularly in adults with minimal or atypical expression of the disease.
Key words
celiac disease, T cells, gluten peptides, HLA-DQ2/DQ8, cytokines, transglutaminase, antibodies
Artículo completo
ASPECTOS INMUNOLOGICOS DE LA ENFERMEDAD CELIACA
(especial para SIIC © Derechos reservados)
Inmunopatogenia de la enfermedad celíaca
La enfermedad celíaca (EC) es un modelo de gran interés para estudiar cómo la interacción entre factores genéticos y ambientales lleva a la pérdida de tolerancia oral a una proteína de la dieta, como es el gluten.1-3 Se ha avanzado mucho en el conocimiento de la patología molecular de la EC, en especial con la identificación de los heterodímeros HLA-DQ2 y DQ8 y su papel en la presentación de gluten a los linfocitos T CD4+ específicos,1 de la acción directa del gluten sobre el epitelio.4-6 La inflamación mucosa y la aparición de la lesión intestinal son secundarios a la activación secuencial de las respuestas inmunitarias innata y adaptativa, que conducen a la alteración de la producción local de citoquinas por los linfocitos T.7-9
El modelo patogénico más aceptado integra factores que actúan tanto en el epitelio como en la lámina propria, como la digestión incompleta y el transporte transepitelial de péptidos,10-12 el efecto tóxico directo del gluten sobre el epitelio, la proliferación y activación de linfocitos intraepiteliales (LIE),6,13 y el reconocimiento de péptidos de gluten por linfocitos T específicos con restricción HLA-DQ2 tras ser modificados por la transglutaminasa tisular (TGt)14,15 (Figura 1). La principal laguna en el conocimiento de la patogenia de la EC es explicar por qué sólo unos pocos individuos portadores del HLA de riesgo desarrollan la enfermedad. Es posible que otros factores no sólo genéticos, sino también ambientales (por ejemplo, la composición de la flora intestinal) influyan en la capacidad individual de inducción y control de la respuesta innata, y en la susceptibilidad del individuo.
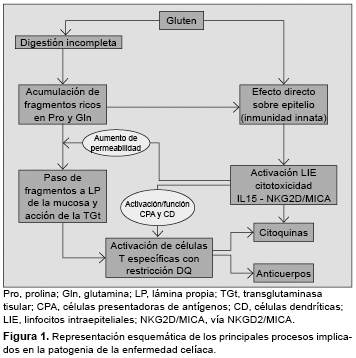
Las principales familias de proteínas del gluten de trigo (gliadinas y gluteninas), y sus homólogos en la cebada y el centeno, denominadas prolaminas por su alto contenido en los aminoácidos glutamina y prolina,16,17 contienen fragmentos nocivos para el intestino celíaco. Se han identificado dos tipos de péptidos, inmunogénicos, que estimulan linfocitos T del intestino o sangre periférica de los pacientes celíacos con restricción DQ2/DQ8, y pueden ser epitopes inmunodominantes (como los residuos 57-75 de alfa-gliadina);14,17-19 y péptidos tóxicos (residuos 31-43/49) de acción directa sobre el epitelio, que es independiente de los linfocitos T.5,7 No todos los cereales contienen la misma proporción de péptidos de cada tipo ni la misma cantidad relativa de gluten, de ahí las variaciones en su capacidad patogénica: la avena tiene alrededor del 10% de contenido de gluten que tiene el trigo.
La EC está fuertemente asociada con genes HLA (locus CELIAC1, cromosoma 6p21): la mayoría de los pacientes celíacos muestran una variante de la molécula HLA-DQ2 codificada por los alelos DQA1*05 y DQB1*02, y el resto son DQ8 (DQA1*03, DQB1*0302), o son portadores de algún alelo aislado del DQ2.20,21 Estos genes muestran un efecto dosis mediado por una presentación de péptidos, que es más eficaz en los homocigotos HLA-DQ2. Aunque el 25% de la población es portadora de DQ2, sólo 1% presenta EC.7,20 Estudios de genoma completo han identificado otras regiones que incluyen genes de susceptibilidad, muchos de ellos relacionados con la función inmunitaria,22 y que podrían ser compartidos también con otras enfermedades crónicas de base inmunológica, como la diabetes mellitus.23
Paso de péptidos de gluten a través del epitelio
La principal forma de entrada de los péptidos de gliadina a través del epitelio es la transcitosis, aunque todavía debe aclararse bien este mecanismo.8 En la EC activa, se ha observado un aumento del transporte transepitelial, y el procesamiento de estos péptidos por las células del epitelio está también alterado, de forma que péptidos tóxicos (19-mer) e inmunogénicos (33-mer), tanto en forma intacta como parcialmente degradada, podrían pasar al interior.10,24 Varios estudios avalan esta posibilidad y se ha observado que en los pacientes celíacos hay un transporte elevado desde la membrana apical de los enterocitos hasta la basal mediante un mecanismo dependiente del interferón gamma (IFN-g).11,12 Además, en un modelo de células Caco-2, la estimulación con IFN-g se asocia con un incremento de la traslocación del péptido 33mer.25
Se ha identificado otro posible mecanismo de transporte transepitelial de la gliadina mediado por el receptor de la transferrina CD71.26 Este receptor está sobreexpresado en la superficie apical de los enterocitos en la EC activa y se une a la IgA secretada para mediar el trasporte de complejos IgA-gliadina. En los pacientes con EC activa se han encontrado también complejos IgG-gliadina. Dado que el receptor neonatal Fc (FcRn) se expresa en las células epiteliales del intestino humano y puede mediar la transcitosis apical a basolateral de inmunocomplejos IgG-antígeno,26 el FcRn podría también transportar inmunocomplejos de IgG antigliadina y gliadina.
Las proteínas de la luz intestinal pueden pasar al interior por transporte paracelular entre los enterocitos. La permeabilidad intestinal está aumentada en los pacientes celíacos por alteración de las uniones estrechas entre los enterocitos. Este hallazgo puede tener un componente genético, ya que se ha observado también en familiares no afectados de los pacientes.27 Sin embargo, por sí misma no explica el tránsito masivo de péptidos que se produce en la EC activa. Otra posibilidad implica un efecto activo de la gliadina sobre la permeabilidad intestinal, que favorecería su debilitamiento. El gluten induciría la secreción paracrina de la proteína zonulina,28 que al ser reconocida por los enterocitos adyacentes dispara una cascada de señalización intracelular que favorece el desacoplamiento de las uniones estrechas entre los enterocitos.25,27,29 La tercera forma de entrada de péptidos podría ser la captación directa por células dendríticas de la lámina propria mucosa que expresan en membrana moléculas HLA-DQ y TG2.30
Inmunidad innata frente al gluten
Péptidos de gluten como el p31-43/49 de la alfa-gliadina pueden dañar directamente el epitelio intestinal al activar mecanismos de la inmunidad innata, con producción de interleuquina (IL) 15. En la EC, hay expresión de IL-15 tanto en los enterocitos del epitelio como en las células mononucleares de la lámina propria mucosa.31,32 La IL-15 favorece la supervivencia, activación y proliferación de los LIE, además de controlar la expansión clonal de los LIE TCR?? y de células con receptores NKG2D,4,33 cuyos ligandos son las moléculas MICA (MHC-I-no clásica) de los enterocitos.31,32,34 Además, la IL-15 favorece la reprogramación tipo-NK de LIE al activar cascadas de señalización intracelular de perforinas/granzimas y de Fas/FasL que contribuyen a desencadenar la inflamación y la citotoxicidad sobre los enterocitos.33,35,36 La IL-15 favorece la retroalimentación de la respuesta inmune al inducir la secreción por los LIE de mediadores de inflamación no específicos, como ácido araquidónico y leucotrienos. También induce la formación del enzima óxido nítrico sintasa inducible (iNOS) por células del estroma de la lámina propria,37,38 mediante un mecanismo dependiente del factor de transcripción NF-kB,27 con el aumento de la permeabilidad y el paso del gluten al interior (Figura 2).
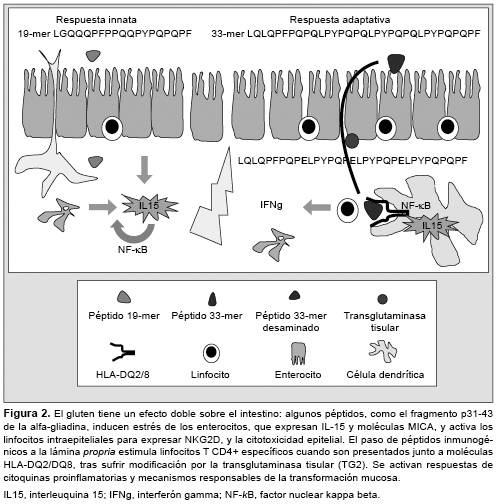
Recientemente se ha conocido que los péptidos de gliadina captados por las células epiteliales mediante endocitosis llegan hasta las vesículas paranucleares (endosomas tardíos y lisosomas), sin embargo, en vez de ser degradados en los lisosomas, el péptido p31-43 se acumula allí por causas aún desconocidas, lo que provoca un microambiente prooxidativo que induce la activación de la TG2 y la degradación de PPAR-gamma (peroxisome proliferator-activated receptor gamma), una molécula capaz de modular la inflamación intestinal. Este mecanismo podría explicar por qué los pacientes celíacos recaen tras la reintroducción del gluten, incluso antes de que aparezcan signos de inflamación.39
Respuesta inmunitaria adaptativa frente al gluten
Los linfocitos T CD4+ específicos de la lámina propria reconocen péptidos sólo cuando son presentados junto a moléculas HLA-DQ2/DQ8 por células dendríticas.15,40 Estas moléculas disponen de un “bolsillo” de unión a péptidos con propiedades únicas para acomodar secuencias peptídicas: DQ2 tiene preferencia por aminoácidos de carga negativa en posiciones centrales (P4, P6, P7), y DQ8, por posiciones más externas (P1, P9).41 De forma natural, las proteínas del gluten tienen pocas cargas negativas, sin embargo, la TG2 liberada durante la inflamación es capaz de inducir la conversión de residuos de glutamina en ácido glutámico en secuencias del tipo QXP (Q = glutamina, P = prolina, X = otro aminoácido)42,43 (Figura 3).
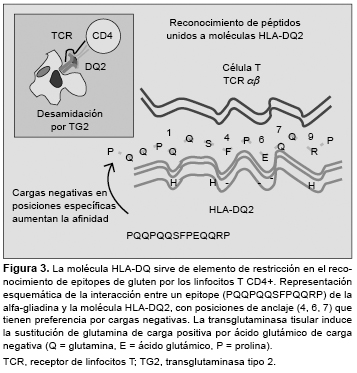
Debido a su alto contenido en glutamina y prolina, los péptidos de gluten son resistentes a la proteólisis por enzimas digestivas, formándose fragmentos grandes que contienen varios motivos QXP, los sustratos preferidos de la TG2,44,45 como el péptido de 33 aminoácidos (p57-89 de la alfa-gliadina), cuya inmunogenicidad para los linfocitos T del intestino celíaco aumenta tras la desamidación por TG2.24 La activación de estos linfocitos T CD4+ reactivos al gluten conduciría a una respuesta proinflamatoria dominada por la producción de IFN-g. Algunas enzimas bacterianas, como la prolil-endopeptidasa, inducen la degradación de estos fragmentos e impiden que la formación de epitopes T activadores de respuestas de la inmunidad adaptativa.46
En la EC en fase activa, hay un aumento del número de células plasmáticas en la lámina propria,47 y la EC se caracteriza por la presencia de anticuerpos séricos frente a distintas moléculas propias y extrañas.48 Los linfocitos B son células presentadoras de antígeno (CPA) específicas. Hay pocos linfocitos B vírgenes o de memoria, y la mayoría son plasmablastos o células plasmáticas con escasa expresión de moléculas HLA-clase II.49 Los linfocitos B podrían jugar un papel importante como CPA en los nódulos linfoides regionales para la amplificación de la respuesta de células T frente al gluten. Los linfocitos B específicos para TG2 estimularían preferentemente los linfocitos T reactivos frente a péptidos de gliadina desamidados, lo que explicaría por qué los anticuerpos frente a estos péptidos son buenos predictores de EC.50
Alteración de la red de citoquinas y mediadores de inflamación
En la EC activa, los linfocitos T CD4+ y los LIE CD8+ desencadenan respuestas Th1 dominadas por IFN-g, el factor de transcripción T bet, y citoquinas proinflamatorias (factor de necrosis tumoral alfa [TNF-a], IL-18, IL-21), junto con la disminución de IL-10 y factor de crecimiento transformante beta (TGF-B),51-53 y la producción de IL-15.5 Este perfil proinflamatorio, que desaparece en la fase de remisión, activa mecanismos efectores del daño tisular. Los fibroblastos del estroma son susceptibles al microambiente local de estrés (óxido nítrico, IFN-g, IL-15, etc.) y secretan el factor de crecimiento de queratinocitos (KGF),54 que podría estar implicado en la hiperplasia de criptas. También se incrementa la expresión de moléculas de adhesión en el endotelio vascular y la síntesis de quimioquinas, que contribuyen al reclutamiento de células inflamatorias, y se estimula la síntesis de metaloproteinasas de la matriz (MMP), una familia de endopeptidasas que pueden degradar componentes de la matriz extracelular (como proteoglucanos y glucoproteínas), y destruir la mucosa.55,56 En la EC se ha descrito una correlación entre los mecanismos de inflamación inespecíficos, como los niveles de expresión de MMP-12, y la presencia de IFN-g; con el grado de lesión mucosa.57
Al contrario de lo que ocurre en otras enfermedades inflamatorias crónicas del intestino, en la EC activa no aumenta la expresión de IL-12, principal citoquina de la diferenciación Th1, por lo que debe haber otras citoquinas que ejerzan esta función, como el IFN-alfa, producido por células dendríticas,58,59 o la IL-21,60,61 cuyo gen ha sido localizado en una región ligada a la susceptibilidad de la EC.22 Las citoquinas producidas por células de la inmunidad adaptativa (IFN-g, IL-21), o innata (IFN-a, IL-15), podrían determinar el desarrollo de la inflamación y la enteropatía,8,61 además de contribuir a la pérdida de tolerancia al gluten por bloqueo de la vía de señalización del TGF-B, por IL-15,62 o por inhibición de la supresión de los linfocitos T efectores por linfocitos T reguladores, a través de la IL-21.8
El NF-kB desempeña también un papel clave en la conexión entre la inmunidad innata y la adaptativa. Las células dendríticas (CD), que inician la inmunidad adaptativa en la lámina propria mediante la presentación antigénica a los linfocitos T CD4+ reactivos al gluten,63 necesitan la activación de NF-kB para aumentar la expresión en membrana de moléculas HLA (DQ2/8) y coestimuladoras (CD80/B7.1, CD86/B7.2, CD83) y, con ello, la función de presentación de antígeno.64 Además, estas células pueden ser activadas por células de la inmunidad innata (NK, iNKT y/o T??) que se activan por las señales de estrés locales.63,65 Por lo tanto, las CD actuarían como un sensor capaz de unir las respuestas innata y adquirida, estimulando también la expansión y función de estas células, además de la producción rápida de perforinas y granzimas, y de ser una fuente de IFN-g.65,66 Ambos bucles de retroalimentación, formados por la interacción entre células de la inmunidad innata/CD y la activación del sistema NFkB/IL-15-iNOS, contribuirían a mantener la situación de estrés en la mucosa intestinal.67
La inmunología en el diagnóstico y el seguimiento de la enfermedad celíaca
Hasta hace poco, las recomendaciones para el diagnóstico de la EC (ESPGHAN 1990) se basaban en la realización, al menos, de una primera biopsia intestinal cuando el paciente tomaba gluten, en algún caso esto es necesario para confirmar definitivamente el diagnóstico la realización de una segunda o incluso una tercera biopsia intestinal y de una prueba de provocación con gluten.68 En las nuevas recomendaciones,69 sin embargo, se establece que en algunos casos de niños sintomáticos y con datos analíticos concluyentes (anticuerpos aTG2 muy elevados junto con anticuerpos AEm positivos y HLA de riesgo) realizados en laboratorios expertos, el especialista podría establecer el diagnóstico de EC sin necesidad de biopsia.
Los resultados de la implementación de estos nuevos criterios deberán ser evaluados de forma prospectiva en cada centro. La biopsia intestinal, además de los estudios de morfometría para confirmar o no la lesión, puede aportar información adicional de utilidad aun en ausencia de lesión atrófica (depósitos de IgA anti-tTG, recuentos de LIE por citometría, etcétera). Hay que tener en cuenta que algunos de los síntomas de la EC en la infancia pueden deberse a otras patologías, y el diagnóstico de los adultos plantea otros problemas, en especial cuando los niveles de anticuerpos séricos son negativos. La sensibilidad de las pruebas serológicas es muy baja cuando la lesión intestinal es leve, aunque sólo el 10% de los casos se relacionan con la ingesta de gluten. Los nuevos criterios tampoco tendrían en cuenta el efecto de la dieta sin gluten, que puede aportar información relevante en los casos dudosos. Finalmente, la presencia de marcadores genéticos de riesgo positivos no implica que la persona vaya a contraer necesariamente la enfermedad.69
Marcadores serológicos de la enfermedad celíaca
Los anticuerpos antigliadina (AAG) no son específicos de la EC, se detectan también en otras enfermedades, e incluso en controles normales. Su relación con la patogenia está por confirmarse, y pueden reflejar el aumento de la permeabilidad intestinal, como sugiere la presencia de anticuerpos contra otras proteínas de la dieta, y su aparición en otras enteropatías.70 Los AAG séricos se elevan en la EC activa, en paralelo con la ingestión de gluten, pero mientras los niveles de AAG-IgA disminuyen rápidamente tras retirar el gluten hasta hacerse negativos, los de clase IgG tienen una cinética más lenta y, en algunos casos, no llegan a desaparecer.71 La edad influye en los niveles de AAG; que son más elevados en los pacientes menores de 2 años, en especial de clase IgA, y su valor diagnóstico disminuye en niños mayores y en los adultos.72
Tanto para el uso aislado de AAG-IgA, como de forma conjunta con AAG IgG, los valores de sensibilidad y especificidad publicados se encuentran entre el 70% y el 80%, aunque hay una gran variabilidad entre los estudios.73 En la actualidad, con los marcadores serológicos alternativos que disponemos, no parece estar justificado el uso de los AAG en el diagnóstico de la EC.74 Las únicas situaciones en las que podría mantenerse la utilización de estos marcadores, son el control de la dieta sin gluten y la falta de sensibilidad de los AEm IgA por debajo de los 2 años de edad, aunque en estos casos, los AAG se ven superados por sus sucesores naturales, los anticuerpos antipéptidos desamidados de gliadina (aDGP).75
La descripción del patrón de anticuerpos antiendomisio (AEm) IgA en la dermatitis herpetiforme y la EC por inmunofluorescencia indirecta (IFI) sobre cortes de esófago de mono,76 o de cordón umbilical humano,77 revolucionó el diagnóstico serológico de la EC, alcanzando cifras de sensibilidad y especificidad superiores al 95%.73 Además de la utilización de los tejidos de mono, el principal problema de estos marcadores es técnico, puesto que la IFI requiere la interpretación del observador, y es por tanto subjetiva, y necesita de un entrenamiento especial y experiencia. Se pueden detectar anticuerpos de clase IgG e IgA, aunque es este último isotipo el que tiene valor diagnóstico en la EC en pacientes competentes para IgA.
Los problemas técnicos de los AEm fueron resueltos con el aislamiento e identificación del principal antígeno del endomisio, la transglutaminasa tisular,78 y las pruebas basadas en la detección de anticuerpos anti-transglutaminasa (aTG2). El uso de antígenos humanos (obtenidos de eritrocitos, o recombinantes) mejoró mucho la especificidad de la prueba, aunque siguen apareciendo algunos falsos positivos en hepatopatías (cirrosis biliar primaria, otras cirrosis hepáticas y hepatitis crónicas por virus C), en la artritis reumatoidea, la psoriasis o la enfermedad de Crohn,79 por lo que en estos casos se aconseja validar los casos positivos para aTG2 con el estudio de AEm. A pesar de todo, los anticuerpos IgA aTG2 tienen valores de sensibilidad cercanos al 100%, y de especificidad entre el 89% y el 96%.73
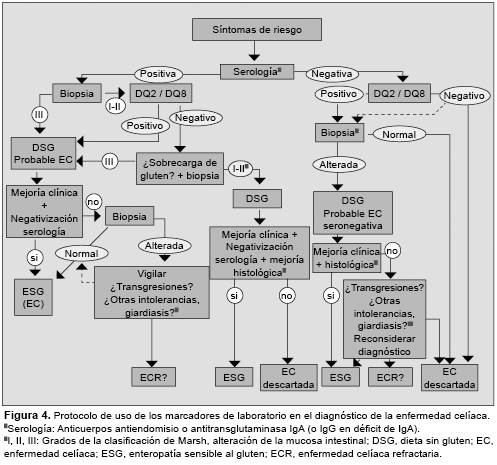
En general, los AEm IgA parecen más específicos y los aTG2 más sensibles, en especial, los que utilizan el antígeno recombinante humano.80 Sin embargo, se ha observado una pérdida de sensibilidad en los casos de lesiones menores en la biopsia, y pueden encontrarse casos de AEm-IgA negativos en un número elevado de los pacientes adultos con este tipo de lesión.81 Un patrón similar, aunque con mejores valores, se ha observado también para los aTGt IgA.82 Por el contrario, pueden encontrarse marcadores positivos sin alteración en biopsia, o con alteraciones leves (Marsh 0 a I).83 Para el control de la dieta, los aTGt IgA pueden ser más adecuados que los AEm IgA,84 aunque hay otras alternativas, como los anticuerpos combinados IgA+IgG aTGt,84,85 o los anticuerpos antipéptidos desaminados de gliadina (aDGP).75 Sin embargo, es difícil determinar cuál es el mejor marcador en el seguimiento de la dieta debido al bajo número de biopsias de control que se realizan (Figura 4).
En los últimos años se han publicado trabajos realizados en población adulta en los que se refleja que el valor de las pruebas serológicas de EC en estos pacientes es mucho menor que en los niños, además de confirmar la existencia de pacientes seronegativos.86 Los estudios in vitro de producción de anticuerpos aTG2 o AEm en cultivos de explantes de biopsias de intestino, podrían resultar de ayuda en estos casos, o evitar las pruebas de provocación in vivo.87 Se han descrito otros autoanticuerpos, como los específicos para proteínas de tipo actina, distintos tipos de colágeno y varios miembros de la familia de la transglutaminasa: TG3, TG6, y factor XIII.88 También se han encontrado complejos formados por IgA/TG3 en la piel de pacientes con dermatitis herpetiforme,50,89 que se asocian con la presencia de anticuerpos frente a la enzima neuronal TG6 en la ataxia.90 Estos hallazgos podrían explicar la aparición de manifestaciones extraintestinales en la EC.
Marcadores inmunogenéticos en la enfermedad celíaca
La asociación de la EC con la región HLA es una de las más fuertes descritas en cualquier patología.91 En la mayoría de las poblaciones estudiadas, más del 90% de los pacientes expresan el heterodímero HLA-DQ2 codificado por los alelos DQA1*05 y DQB1*02 en la posición cis, asociados a DR3 (más común en el centro y norte de Europa), o en trans, en heterocigotos DR5/DR7 (que es más frecuente en la cuenca del Mediterráneo.) Sin embargo, DQ2 está presente en alrededor del 30% de la población general.92 Del resto de los pacientes, muchos presentan un segundo heterodímero de riesgo DQ8 (codificado por los alelos DQA1*03 y DQB1*0302, en cis y asociado al DR4).93 Los pacientes que no expresan ni DQ2 ni DQ8 completos son portadores de algunos de los alelos que codifican para el DQ2 por separado (DQA1*05 o DQB1*02).94 Entre los individuos portadores del DQ2, los que presentan una doble dosis del alelo DQB1*02 podrían tener un mayor riesgo de EC, como es el caso de los homocigotos DR3/DR3 y los heterocigotos DR3/DR7.95,96
Otros alelos de riesgo en la región HLA
La concordancia del 30% entre hermanos con HLA idéntico sugiere que otros genes de dentro y de fuera de la región HLA intervienen en la susceptibilidad. Además, menos del 2% de los portadores del DQ2 o del DQ8 evolucionan a EC, por lo que es probable la acumulación de riesgos debidos a otros muchos genes de acción menor, que podrían ser distintos para cada población o para cada individuo. La mayoría de los pacientes DQ2 positivos son portadores del haplotipo ancestral (AH) 8.1 (B8-DR3-DQ2),97 que incluye otros alelos capaces de conferir riesgo o de modificar el efecto del DQ2.98 Este haplotipo está asociado con otras enfermedades autoinmunitarias. Entre los posibles genes con implicación funcional incluidos en la región HLA estarían los genes del TNF-a99,100 y de la linfotoxina alfa (LT-a). Se han encontrado asociaciones con otros genes de la región HLA, como los que codifican las moléculas MIC-A y MIC-B,101,102 y moléculas de la familia de las proteínas de estrés HSP-70.103 La falta de replicación de estos hallazgos podría deberse a diferencias poblacionales en la contribución a la susceptibilidad.3
Asociación con otras regiones genéticas distintas del HLA
Otras zonas del genoma contienen también genes candidatos que podrían participar en la susceptibilidad a la EC, como las localizadas en 2p33: CELIAC3 (OMIM #609755), 5q31-33: CELIAC2 (OMIM #609754), 15q11-13: CELIAC5 (OMIM %607202) y 19p13.1: CELIAC4 (OMIM #609753). Estas zonas han sido definidas mediante estudios de análisis de ligamiento sistemáticos pangenómicos. Los estudios de asociación son el abordaje para comprobar la implicación de genes candidatos en zonas “calientes” que, en la mayoría, contienen genes relacionados con la respuesta inmunitaria. En cada paciente, se postula que diferentes combinaciones de las variantes de genes de efecto menor podrían determinar el curso y/o la expresión de la EC.104,105
En un estudio de búsqueda pangenómica mediante una técnica de array de SNP con rastreo de más de 300 000 polimorfismos en una amplia muestra de pacientes celíacos y controles,22 se encontró que la única zona del genoma (aparte del HLA) con asociación significativa se localizaba en 4q27, en una zona en la que se encuentran los genes de la IL-2 y la IL-21. Además, se observó un aumento de la expresión del ARNm de IL-21 en un grupo de pacientes con EC activa, en comparación con los controles. Posteriormente, sumando al estudio inicial varias colecciones de ADN europeas (1 643 pacientes y 3 406 controles), se pudo definir varios genes de moléculas relacionadas con el sistema inmunitario como posibles factores de riesgo de implicación funcional: CCR3, IL12A, IL18RAP, RGS1, SH2B3 y TAGAP, algunas compartidas también por la susceptibilidad a la diabetes mellitus tipo 1.106 Sin embargo, en este momento, sólo la IL-21 podría tener un papel en la producción de la lesión mucosa.60
En el último estudio de asociación pangenómica con la EC realizado en población europea se validan parte de los hallazgos anteriores, y se encuentran nuevas regiones de asociación. En general, los posibles genes candidatos se agruparán en cuatro categorías funcionales: desarrollo de linfocitos T en el timo; detección vírica por el sistema inmunitario innato; activación de los linfocitos B y T, y citoquinas, quimioquinas y sus receptores. Sin embargo, se observa una gran variabilidad entre poblaciones al discriminar los resultados de acuerdo con su procedencia.107 Sin embargo, queda por determinar qué papel tienen estas variaciones genéticas en el fenotipo inmunológico característico de la EC.3
Nuevas técnicas en el diagnóstico en la enfermedad celíaca
Inmunofenotipificación por citometría de flujo: el linfograma intraepitelial
Los cambios en las subpoblaciones de LIE son una característica constante en la mucosa duodenal de los pacientes con EC, por lo que su caracterización mediante citometría de flujo es una herramienta diagnóstica de utilidad. Estas variaciones son las siguientes: incremento del porcentaje total de LIE (en relación con el total de células del epitelio) que se observa durante las fases activas de la enfermedad y se normaliza tras la exclusión del gluten de la dieta; reducción de la fracción de células CD3- respecto del total de LIE, que llega a alcanzar valores prácticamente indetectables, normalmente < 20%, y que sufre también una recuperación tras la exclusión del gluten de la dieta;108 aumento del número de células T?d+ respecto al total de LIE.109,110 Este aumento se considera casi patognomónico de la EC y permanece constante tras la retirada del gluten. El valor medio que suele alcanzar esta población se sitúa en torno del 25%.111 Este aumento se observa en todas las fases de la EC, es permanente e intenso, a diferencia del aumento observado en otras enteropatías como intolerancia alimentaria, criptosporidiasis, giardiasis, o déficit de IgA,109 donde el aumento es transitorio y moderado.
Las ventajas de la citometría de flujo como herramienta de diagnóstico en la EC son: 1) complementar el estudio anatomopatológico convencional aumentando su especificidad, ya que se realiza al mismo tiempo con una sola pieza endoscópica; 2) las alteraciones observadas en las subpoblaciones de LIE parecen ser extensivas a las formas latentes de la enfermedad y a la forma de expresión cutánea de la sensibilidad al gluten (dermatitis herpetiforme), en las que tanto las alteraciones anatomopatológicas como los marcadores séricos son de aparición inconstante;112 3) el incremento de las células T?d permanece constante tras la exclusión del gluten, lo cual evita la realización de pruebas de provocación con gluten para confirmar el diagnóstico de EC, y 4) la persistencia de un número elevado de LIE en la biopsia de control tras el tratamiento de exclusión ayuda en la detección de transgresiones dietéticas.113
Detección de péptidos inmunotóxicos de la gliadina en heces
En los últimos años se han realizado ensayos con el fin de desarrollar anticuerpos de utilidad en la detección de gluten, en especial, aplicados al ámbito de la industria alimentaria. Muchos de ellos no son específicos frente a los péptidos inmunotóxicos de la gliadina, por lo que su utilización podría dar lugar a falsos positivos, en los que la detección de fragmentos del gluten no conlleva necesariamente la presencia de péptidos inmunotóxicos. Sin embargo, se han identificado anticuerpos, como los denominados G12 y A1 (Biomedal S.L., Sevilla, España) que son específicos para el péptido inmunotóxico 33mer, y que han demostrado ser eficaces para evaluar la seguridad de los alimentos libres de gluten.114
Estudios publicados recientemente por Comino y colaboradores115 han permitido desarrollar un novedoso método para la detección y monitorización de la presencia de péptidos del gluten de carácter inmunodominante en muestras de heces de pacientes con EC, utilizando el anticuerpo G12 anti-33mer en una prueba de ELISA. La resistencia a la digestión gastrointestinal que presentan diversos péptidos derivados del gluten, y en particular el 33mer, asegura que una parte significativa del gluten ingerido será excretado en heces en forma de fracciones peptídicas no digeridas. La detección de fracciones proteicas inmunotóxicas en heces indica que el gluten ha sido ingerido y ha atravesado el tracto digestivo, confirmándose la existencia de una correlación entre la cantidad de gluten consumida y la cantidad de gluten excretada.
Actualmente no existen métodos válidos para monitorizar el cumplimiento correcto de la dieta sin gluten. Se propuso la determinación de anticuerpos aTG como un buen marcador, sin embargo, la recuperación de los valores normales tras eliminar el gluten de la dieta puede durar meses e incluso años.116,117 La utilización de endoscopias seriadas sería determinante, pero no puede ser considerada como una práctica clínica ética.118,119 Las entrevistas dietéticas pueden ser útiles para determinar el grado de cumplimiento de la dieta, aunque no están bien estandarizadas, son subjetivas, dependen de la respuesta verdadera del paciente y no identifican transgresiones involuntarias. La cuantificación de péptidos de gliadina (como 33mer) en heces mediante anticuerpos específicos permitiría detectar el consumo de gluten de una forma fiable y no invasiva, y con ello monitorizar el correcto cumplimiento de la dieta sin gluten a corto plazo, estudiar la eficacia de las terapias enzimáticas que se encuentran actualmente en desarrollo, e identificar transgresiones involuntarias de la dieta, relacionadas con contaminaciones de productos aparentemente sin gluten.115
La inmunología en el desarrollo de nuevas terapias en la enfermedad celíaca
Hasta el momento, el único tratamiento efectivo y seguro de la EC es la dieta estricta sin gluten, de por vida. Sin embargo, es conocido que el seguimiento de esta dieta no siempre es fácil, tiene un costo económico más elevado y, además, el gluten de trigo es un componente esencial de la alimentación y se utiliza frecuentemente como aditivo. Sea de forma voluntaria o involuntaria, las transgresiones dietéticas pueden desencadenar la cronificación de la enfermedad, así como la aparición de complicaciones graves asociadas.
Hasta la fecha se han realizado ensayos clínicos con cinco alternativas terapéuticas, potencialmente complementarias entre sí. El acetato de larazotido AT-1001, desarrollado por la compañía americana Alba Therapeutics, tiene como objetivo impedir el paso paracelular del gluten a la lámina propria mediante reordenación y cierre de las uniones intercelulares densas.120 Las endopeptidasas (ALV003 y AN-PEP) degradarían el gluten hasta conseguir fragmentos no tóxicos que, al alcanzar la lámina propria, no desencadenarían una respuesta inmunitaria patológica.117,121 Por otro lado, se están ensayando vacunas terapéuticas que intentan conseguir tolerancia al gluten (NexVax2),124 además de los estudios basados en la infección con parásitos como Necator americanus,122 y la utilización de fármacos inmunomoduladores, como el inhibidor de los receptores CCR9 de los linfocitos T (CCX282-B, Traficet-EN).123
|
Bibliografía del artículo
1. Sollid LM. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol 2(9):647-55, 2002.
2. Jabri B, Sollid LM. Tissue-mediated control of immunopathology in coeliac disease. Nat Rev Immunol 9(12):858-70, 2009.
3. Abadie V, Sollid LM, Barreiro LB, Jabri B. Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annu Rev Immunol 29:493-525, 2011.
4. Hue S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity 21(3):367-77, 2004.
5. Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Auricchio S, et al. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 362(9377):30-7, 2003.
6. Meresse B, Curran SA, Ciszewski C, Orbelyan G, Setty M, Bhagat G, et al. Reprogramming of CTLs into natural killer-like cells in celiac disease. J Exp Med 203(5):1343-55, 2006.
7. Jabri B, Sollid LM. Mechanisms of disease: immunopathogenesis of celiac disease. Nat Clin Pract Gastroenterol Hepatol 3(9):516-25, 2006.
8. Meresse B, Ripoche J, Heyman M, Cerf-Bensussan N. Celiac disease: from oral tolerance to intestinal inflammation, autoimmunity and lymphomagenesis. Mucosal Immunol 2(1):8-23, 2009.
9. Tjon JM, Van Bergen J, Koning F. Celiac disease: how complicated can it get? Immunogenetics 62(10):641-51, 2010.
10. Matysiak-Budnik T, Candalh C, Dugave C, Namane A, Cellier C, Cerf-Bensussan N, et al. Alterations of the intestinal transport and processing of gliadin peptides in celiac disease. Gastroenterology 125(3):696-707, 2003.
11. Zimmer KP, Fischer I, Mothes T, Weissen-Plenz G, Schmitz M, Wieser H, et al. Endocytotic segregation of gliadin peptide 31-49 in enterocytes. Gut 59(3):300-10, 2010.
12. Luciani A, Villella VR, Vasaturo A, Giardino I, Pettoello-Mantovani M, Guido S, et al. Lysosomal accumulation of gliadin p31-43 peptide induces oxidative stress and tissue transglutaminase-mediated PPARgamma downregulation in intestinal epithelial cells and coeliac mucosa. Gut 59(3):311-9, 2010.
13. Abadie V, Discepolo V, Jabri B. Intraepithelial lymphocytes in celiac disease immunopathology. Semin Immunopathol 34(4):551-66, 2011.
14. Kagnoff MF. Celiac disease: pathogenesis of a model immunogenetic disease. J Clin Invest 117(1):41-9, 2007.
15. Sollid LM. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol 2(9):647-55, 2002.
16. Shewry PR, Halford NG. Cereal seed storage proteins: structures, properties and role inthe grain utilization. J Exp Bot 53:947-58, 2003.
17. Sollid LM, Qiao SW, Anderson RP, Gianfrani C, Koning F. Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules. Immunogenetics 64(6):455-60, 2011.
18. Arentz-Hansen H, McAdam SN, Molberg O, Fleckenstein B, Lundin KE, Jorgensen TJ, et al. Celiac lesion T cells recognize epitopes that cluster in regions of gliadins rich in proline residues. Gastroenterology 123(3):803-9, 2002.
19. Vader W, Kooy Y, Van Veelen P, De Ru A, Harris D, Benckhuijsen W, et al. The gluten response in children with celiac disease is directed toward multiple gliadin and glutenin peptides. Gastroenterology 122(7):1729-37, 2002.
20. Sollid LM, Lie BA. Celiac disease genetics: current concepts and practical applications. Clin Gastroenterol Hepatol 3(9):843-51, 2005.
21. Sollid LM, Thorsby E. HLA susceptibility genes in celiac disease: genetic mapping and role in pathogenesis. Gastroenterology 105(3):910-22, 1993.
22. Van Heel DA, Franke L, Hunt KA, Gwilliam R, Zhernakova A, Inouye M, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet 39(7):827-9, 2007.
23. Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JH, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med 359(26):2767-77, 2008.
24. Shan L, Molberg O, Parrot I, Hausch F, Filiz F, Gray GM, et al. Structural basis for gluten intolerance in celiac sprue. Science 297(5590):2275-9, 2002.
25. Menard S, Lebreton C, Schumann M, Matysiak-Budnik T, Dugave C, Bouhnik Y, et al. Paracellular versus transcellular intestinal permeability to gliadin peptides in active celiac disease. Am J Pathol 180(2):608-15, 2012.
26. Matysiak-Budnik T, Candalh C, Dugave C, Namane A, Cellier C, Cerf-Bensussan N, et al. Alterations of the intestinal transport and processing of gliadin peptides in celiac disease. Gastroenterology 125(3):696-707, 2003.
27. Clemente MG, De Virgiliis S, Kang JS, Macatagney R, Musu MP, Di Pierro MR, et al. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut 52(2):218-23, 2003.
28. Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology 135(1):194-204 e3, 2008.
29. Drago S, El Asmar R, Di Pierro M, Grazia Clemente M, Tripathi A, Sapone A, et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol 41(4):408-19, 2006.
30. Raki M, Tollefsen S, Molberg O, Lundin KE, Sollid LM, Jahnsen FL. A unique dendritic cell subset accumulates in the celiac lesion and efficiently activates gluten-reactive T cells. Gastroenterology 131(2):428-38, 2006.
31. Maiuri L, Ciacci C, Auricchio S, Brown V, Quaratino S, Londei M. Interleukin 15 mediates epithelial changes in celiac disease. Gastroenterology 119(4):996-1006, 2000.
32. Mention JJ, Ben Ahmed M, Begue B, Barbe U, Verkarre V, Asnafi V, et al. Interleukin 15: a key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology 125(3):730-45, 2003.
33. Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 21(3):357-66, 2004.
34. Maiuri L, Ciacci C, Vacca L, Ricciardelli I, Auricchio S, Quaratino S, et al. IL-15 drives the specific migration of CD94 and TCR-gammadelta intraepithelial lymphocytes in organ cultures of treated celiac patients. Am J Gastroenterol 96(1):150-6, 2001.
35. Di Sabatino A, Ciccocioppo R, Cupelli F, Cinque B, Millimaggi D, Clarkson MM, et al. Epithelium derived interleukin 15 regulates intraepithelial lymphocyte Th1 cytokine production, cytotoxicity, and survival in coeliac disease. Gut 55(4):469-77, 2006.
36. Ebert EC. IL-15 converts human intestinal intraepithelial lymphocytes to CD94 producers of IFN-gamma and IL-10, the latter promoting Fas ligand-mediated cytotoxicity. Immunology 115(1):118-26, 2005.
37. Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Auricchio S, et al. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 362(9377):30-7, 2003.
38. De Stefano D, Maiuri MC, Iovine B, Ialenti A, Bevilacqua MA, Carnuccio R. The role of NF-kappaB, IRF-1, and STAT-1alpha transcription factors in the iNOS gene induction by gliadin and IFN-gamma in RAW 264.7 macrophages. J Mol Med (Berl) 84(1):65-74, 2006.
39. Luciani A, Villella VR, Vasaturo A, Giardino I, Pettoello-Mantovani M, Guido S, et al. Lysosomal accumulation of gliadin p31-43 peptide induces oxidative stress and Tissue Transglutaminase mediated PPAR downregulation in intestinal epithelial cells and coeliac mucosa. Gut 59(7):1007, 2010.
40. Lundin KE, Scott H, Hansen T, Paulsen G, Halstensen TS, Fausa O, et al. Gliadin-specific, HLA-DQ(alpha 1*0501,beta 1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. J Exp Med 178(1):187-96, 1993.
41. Van de Wal Y, Kooy Y, Van Veelen P, Pena S, Mearin L, Papadopoulos G, et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol 161(4):1585-8, 1998.
42. Fleckenstein B, Qiao SW, Larsen MR, Jung G, Roepstorff P, Sollid LM. Molecular characterization of covalent complexes between tissue transglutaminase and gliadin peptides. J Biol Chem 279(17):17607-16, 2004.
43. Molberg O, McAdam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med 4(6):713-7, 1998.
44. Arentz-Hansen H, Korner R, Molberg O, Quarsten H, Vader W, Kooy YM, et al. The intestinal T cell response to alpha-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J Exp Med 191(4):603-12, 2000.
45. Hausch F, Shan L, Santiago NA, Gray GM, Khosla C. Intestinal digestive resistance of immunodominant gliadin peptides. Am J Physiol Gastrointest Liver Physiol 283(4):G996-G1003, 2002.
46. Piper JL, Gray GM, Khosla C. Effect of prolyl endopeptidase on digestive-resistant gliadin peptides in vivo. J Pharmacol Exp Ther 311(1):213-9, 2004.
47. Farstad IN, Halstensen TS, Kvale D, Fausa O, Brandtzaeg P. Topographic distribution of homing receptors on B and T cells in human gut-associated lymphoid tissue: relation of L-selectin and integrin alpha 4 beta 7 to naive and memory phenotypes. Am J Pathol 150(1):187-99, 1997.
48. Dieterich W, Storch WB, Schuppan D. Serum antibodies in celiac disease. Clin Lab 46(7-8):361-4, 2000.
49. Farstad IN, Carlsen H, Morton HC, Brandtzaeg P. Immunoglobulin A cell distribution in the human small intestine: phenotypic and functional characteristics. Immunology 101(3):354-63, 2000.
50. Qiao SW, Iversen R, Raki M, Sollid LM. The adaptive immune response in celiac disease. Semin Immunopathol 34(4):523-40, 2012.
51. Forsberg G, Hernell O, Melgar S, Israelsson A, Hammarstrom S, Hammarstrom ML. Paradoxical coexpression of proinflammatory and down-regulatory cytokines in intestinal T cells in childhood celiac disease. Gastroenterology 123(3):667-78, 2002.
52. Leon AJ, Garrote JA, Blanco-Quiros A, Calvo C, Fernandez-Salazar L, Del Villar A, et al. Interleukin 18 maintains a long-standing inflammation in coeliac disease patients. Clin Exp Immunol 146(3):479-85, 2006.
53. Nilsen EM, Lundin KE, Krajci P, Scott H, Sollid LM, Brandtzaeg P. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut 37(6):766-76, 1995.
54. Salvati VM, Bajaj-Elliott M, Poulsom R, Mazzarella G, Lundin KE, Nilsen EM, et al. Keratinocyte growth factor and coeliac disease. Gut 49(2):176-81, 2001.
55. Macdonald TT, Monteleone G. Immunity, inflammation, and allergy in the gut. Science 307(5717):1920-5, 2005.
56. Pender SL, MacDonald TT. Matrix metalloproteinases and the gut - new roles for old enzymes. Curr Opin Pharmacol 4(6):546-50, 2004.
57. Ciccocioppo R, Di Sabatino A, Bauer M, Della Riccia DN, Bizzini F, Biagi F, et al. Matrix metalloproteinase pattern in celiac duodenal mucosa. Lab Invest 85(3):397-407, 2005.
58. Di Sabatino A, Pickard KM, Gordon JN, Salvati V, Mazzarella G, Beattie RM, et al. Evidence for the role of interferon-alfa production by dendritic cells in the Th1 response in celiac disease. Gastroenterology 133(4):1175-87, 2007.
59. Monteleone G, Pender SL, Alstead E, Hauer AC, Lionetti P, McKenzie C, et al. Role of interferon alpha in promoting T helper cell type 1 responses in the small intestine in coeliac disease. Gut 48(3):425-9, 2001.
60. Fina D, Sarra M, Caruso R, Del Vecchio Blanco G, Pallone F, Macdonald TT, et al. Interleukin-21 contributes to the mucosal t helper cell type 1 response in celiac disease. Gut 57(7):887-92, 2008.
61. Garrote JA, Gomez-Gonzalez E, Bernardo D, Arranz E, Chirdo F. Celiac disease pathogenesis: the proinflammatory cytokine network. J Pediatr Gastroenterol Nutr 47(Suppl 1):S27-32, 2008.
62. Benahmed M, Meresse B, Arnulf B, Barbe U, Mention JJ, Verkarre V, et al. Inhibition of TGF-beta signaling by IL-15: a new role for IL-15 in the loss of immune homeostasis in celiac disease. Gastroenterology 132(3):994-1008, 2007.
63. Munz C, Steinman RM, Fujii S. Dendritic cell maturation by innate lymphocytes: coordinated stimulation of innate and adaptive immunity. J Exp Med 202(2):203-7, 2005.
64. Calder VL, Bondeson J, Brennan FM, Foxwell BM, Feldmann M. Antigen-specific T-cell downregulation by human dendritic cells following blockade of NF-kappaB. Scand J Immunol 57(3):261-70, 2003.
65. Yu KO, Porcelli SA. The diverse functions of CD1d-restricted NKT cells and their potential for immunotherapy. Immunol Lett 100(1):42-55, 2005.
66. Van der Vliet HJ, Molling JW, Von Blomberg BM, Nishi N, Kolgen W, Van den Eertwegh AJ, et al. The immunoregulatory role of CD1d-restricted natural killer T cells in disease. Clin Immunol 112(1):8-23, 2004.
67. Stepniak D, Koning F. Celiac disease--sandwiched between innate and adaptive immunity. Hum Immunol 67(6):460-8, 2006.
68. Ribes-Koninckx C, Mearin ML, Korponay-Szabo IR, Shamir R, Husby S, Ventura A, et al. Coeliac disease diagnosis: ESPGHAN 1990 criteria or need for a change? Results of a questionnaire. J Pediatr Gastroenterol Nutr 54(1):15-9, 2011.
69. Husby S, Koletzko S, Korponay-Szabo IR, Mearin ML, Phillips A, Shamir R, et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr 54(1):136-60, 2012.
70. Ferguson A, Carswell F. Precipitins to dietary proteins in serum and upper intestinal secretions of coeliac children. Br Med J 1(5792):75-7, 1972.
71. Signer E, Burgin-Wolff A, Berger R, Birbaumer A, Just M. Antibodies to gliadin as a screening test for coeliac disease. A prospective study. Helv Paediatr Acta 34(1):41-52, 1979.
72. Savilahti E, Viander M, Perkkio M, Vainio E, Kalimo K, Reunala T. IgA antigliadin antibodies: a marker of mucosal damage in childhood coeliac disease. Lancet 1(8320):320-2, 1983.
73. Rostom A, Dube C, Cranney A, Saloojee N, Sy R, Garritty C, et al. The diagnostic accuracy of serologic tests for celiac disease: a systematic review. Gastroenterology 128(4 Suppl 1):S38-46, 2005.
74. Green PH, Rostami K, Marsh MN. Diagnosis of coeliac disease. Best Pract Res Clin Gastroenterol 19(3):389-400, 2005.
75. Agardh D. Antibodies against synthetic deamidated gliadin peptides and tissue transglutaminase for the identification of childhood celiac disease. Clin Gastroenterol Hepatol 5(11):1276-81, 2007.
76. Chorzelski TP, Beutner EH, Sulej J, Tchorzewska H, Jablonska S, Kumar V, et al. IgA anti-endomysium antibody. A new immunological marker of dermatitis herpetiformis and coeliac disease. Br J Dermatol 111(4):395-402, 1984.
77. Amara W, Husebekk A. Improved method for serological testing in celiac disease--IgA anti-endomysium antibody test: a comparison between monkey oesophagus and human umbilical cord as substrate in indirect immunofluorescence test. Scand J Clin Lab Invest 58(7):547-54, 1998.
78. Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med 3(7):797-801, 1997.
79. Carroccio A, Vitale G, Di Prima L, Chifari N, Napoli S, La Russa C, et al. Comparison of anti-transglutaminase ELISAs and an anti-endomysial antibody assay in the diagnosis of celiac disease: a prospective study. Clin Chem 48(9):1546-50, 2002.
80. Lewis NR, Scott BB. Systematic review: the use of serology to exclude or diagnose coeliac disease (a comparison of the endomysial and tissue transglutaminase antibody tests). Aliment Pharmacol Ther 24(1):47-54, 2006.
81. Rostami K, Kerckhaert JP, Tiemessen R, Meijer JW, Mulder CJ. The relationship between anti-endomysium antibodies and villous atrophy in coeliac disease using both monkey and human substrate. Eur J Gastroenterol Hepatol 11(4):439-42, 1999.
82. Abrams JA, Brar P, Diamond B, Rotterdam H, Green PH. Utility in clinical practice of immunoglobulin a anti-tissue transglutaminase antibody for the diagnosis of celiac disease. Clin Gastroenterol Hepatol 4(6):726-30, 2006.
83. Hadithi M, Von Blomberg BM, Crusius JB, Bloemena E, Kostense PJ, Meijer JW, et al. Accuracy of serologic tests and HLA-DQ typing for diagnosing celiac disease. Ann Intern Med 147(5):294-302, 2007.
84. Bazzigaluppi E, Roggero P, Parma B, Brambillasca MF, Meroni F, Mora S, et al. Antibodies to recombinant human tissue-transglutaminase in coeliac disease: diagnostic effectiveness and decline pattern after gluten-free diet. Dig Liver Dis 38(2):98-102, 2006.
85. Reeves GE, Squance ML, Duggan AE, Murugasu RR, Wilson RJ, Wong RC, et al. Diagnostic accuracy of coeliac serological tests: a prospective study. Eur J Gastroenterol Hepatol 18(5):493-501, 2006.
86. Abrams JA, Diamond B, Rotterdam H, Green PH. Seronegative celiac disease: increased prevalence with lesser degrees of villous atrophy. Dig Dis Sci 49(4):546-50, 2004.
87. Bonamico M, Sabbatella L, Di Tola M, Vetrano S, Ferri M, Nenna R, et al. Antiendomysial antibody detection in biopsy culture allows avoidance of gluten challenge in celiac children. J Pediatr Gastroenterol Nutr 40(2):165-9, 2005.
88. Alaedini A, Green PH. Autoantibodies in celiac disease. Autoimmunity 41(1):19-26, 2008.
89. Sardy M, Karpati S, Merkl B, Paulsson M, Smyth N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J Exp Med 195(6):747-57, 2002.
90. Hadjivassiliou M, Aeschlimann P, Strigun A, Sanders DS, Woodroofe N, Aeschlimann D. Autoantibodies in gluten ataxia recognize a novel neuronal transglutaminase. Ann Neurol 64(3):332-43, 2008.
91. Louka AS, Sollid LM. HLA in coeliac disease: unravelling the complex genetics of a complex disorder. Tissue Antigens 61(2):105-17, 2003.
92. Arranz E, Garrote JA. HLA en la enfermedad celiaca. An Pediatr Contin 2(3):163-6, 2004.
93. Karell K, Louka AS, Moodie SJ, Ascher H, Clot F, Greco L, et al. Hla types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the European Genetics Cluster on Celiac Disease. Hum Immunol 64(4):469-77, 2003.
94. Polvi A, Arranz E, Fernandez-Arquero M, Collin P, Maki M, Sanz A, et al. HLA-DQ2-negative celiac disease in Finland and Spain. Hum Immunol 59(3):169-75, 1998.
95. Arranz E, Telleria JJ, Sanz A, Martin JF, Alonso M, Calvo C, et al. HLA-DQA1*0501 and DQB1*02 homozygosity and disease susceptibility in Spanish coeliac patients. Exp Clin Immunogenet 14(4):286-90, 1997.
96. Bourgey M, Calcagno G, Tinto N, Gennarelli D, Margaritte-Jeannin P, Greco L, et al. HLA related genetic risk for coeliac disease. Gut 56(8):1054-9, 2007.
97. Candore G, Lio D, Colonna Romano G, Caruso C. Pathogenesis of autoimmune diseases associated with 8.1 ancestral haplotype: effect of multiple gene interactions. Autoimmun Rev 1(1-2):29-35, 2002.
98. Louka AS, Moodie SJ, Karell K, Bolognesi E, Ascher H, Greco L, et al. A collaborative European search for non-DQA1*05-DQB1*02 celiac disease loci on HLA-DR3 haplotypes: analysis of transmission from homozygous parents. Hum Immunol 64(3):350-8, 2003.
99. Garrote JA, Arranz E, Telleria JJ, Castro J, Calvo C, Blanco-Quiros A. TNF alpha and LT alpha gene polymorphisms as additional markers of celiac disease susceptibility in a DQ2-positive population. Immunogenetics 54(8):551-5, 2002.
100. Louka AS, Lie BA, Talseth B, Ascher H, Ek J, Gudjonsdottir AH, et al. Coeliac disease patients carry conserved HLA-DR3-DQ2 haplotypes revealed by association of TNF alleles. Immunogenetics 55(5):339-43, 2003.
101. Lopez-Vazquez A, Fuentes D, Rodrigo L, Gonzalez S, Moreno M, Fernandez E, et al. MHC class I region plays a role in the development of diverse clinical forms of celiac disease in a Saharawi population. Am J Gastroenterol 99(4):662-7, 2004.
102. Bilbao JR, Martin-Pagola A, Perez De Nanclares G, Calvo B, Vitoria JC, Vazquez F, et al. HLA-DRB1 and MICA in autoimmunity: common associated alleles in autoimmune disorders. Ann N Y Acad Sci 1005:314-8, 2003.
103. Ramos-Arroyo MA, Feijoo E, Sanchez-Valverde F, Aranburu E, Irisarri N, Olivera JE, et al. Heat-shock protein 70-1 and HLA class II gene polymorphisms associated with celiac disease susceptibility in Navarra (Spain). Hum Immunol 62(8):821-5, 2001.
104. Monsuur AJ, De Bakker PI, Alizadeh BZ, Zhernakova A, Bevova MR, Strengman E, et al. Myosin IXB variant increases the risk of celiac disease and points toward a primary intestinal barrier defect. Nat Genet 37(12):1341-4, 2005.
105. Holopainen P, Naluai AT, Moodie S, Percopo S, Coto I, Clot F, et al. Candidate gene region 2q33 in European families with coeliac disease. Tissue Antigens 63(3):212-22, 2004.
106. Hunt KA, Zhernakova A, Turner G, Heap GA, Franke L, Bruinenberg M, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet 40(4):395-402, 2008.
107. Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet 42(4):295-302, 2010.
108. Eiras P, Leon F, Camarero C, Lombardia M, Roldan E, Bootello A, et al. Intestinal intraepithelial lymphocytes contain a CD3- CD7 subset expressing natural killer markers and a singular pattern of adhesion molecules. Scand J Immunol 52(1):1-6, 2000.
109. Spencer J, Isaacson PG, MacDonald TT, Thomas AJ, Walker-Smith JA. Gamma/delta T cells and the diagnosis of coeliac disease. Clin Exp Immunol 85(1):109-13, 1991.
110. Arranz E, Bode J, Kingstone K, Ferguson A. Intestinal antibody pattern of coeliac disease: association with gamma/delta T cell receptor expression by intraepithelial lymphocytes, and other indices of potential coeliac disease. Gut 35(4):476-82, 1994.
111. Leon F, Eiras P, Camarero C, Roldan E, Sanchez L, R RP, et al. Advances in the diagnosis of celiac disease: anti-transglutaminase antibodies and intestinal intraepithelial lymphocytes. Gastroenterol Hepatol 25(6):416-22, 2002.
112. Savilahti E, Ormala T, Arato A, Hacsek G, Holm K, Klemola T, et al. Density of gamma/delta T cells in the jejunal epithelium of patients with coeliac disease and dermatitis herpetiformis is increased with age. Clin Exp Immunol 109(3):464-7, 1997.
113. Leon F. Flow cytometry of intestinal intraepithelial lymphocytes in celiac disease. J Immunol Methods 363(2):177-86, 2011.
114. Moron B, Bethune MT, Comino I, Manyani H, Ferragud M, Lopez MC, et al. Toward the assessment of food toxicity for celiac patients: characterization of monoclonal antibodies to a main immunogenic gluten peptide. PLoS One 3(5):e2294, 2008.
115. Comino I, Real A, Vivas S, Siglez MA, Caminero A, Nistal E, et al. Monitoring of gluten-free diet compliance in celiac patients by assessment of gliadin 33-mer equivalent epitopes in feces. Am J Clin Nutr 95(3):670-7, 2012.
116. Walker MM, Murray JA. An update in the diagnosis of coeliac disease. Histopathology 59(2):166-79, 2011.
117. Tack GJ, Verbeek WH, Schreurs MW, Mulder CJ. The spectrum of celiac disease: epidemiology, clinical aspects and treatment. Nat Rev Gastroenterol Hepatol 7(4):204-13, 2010.
118. Duerksen DR, Wilhelm-Boyles C, Parry DM. Intestinal permeability in long-term follow-up of patients with celiac disease on a gluten-free diet. Dig Dis Sci 50(4):785-90, 2005.
119. Ertekin V, Selimoglu MA, Turgut A, Bakan N. Fecal calprotectin concentration in celiac disease. J Clin Gastroenterol 44(8):544-6, 2010.
120. Paterson BM, Lammers KM, Arrieta MC, Fasano A, Meddings JB. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther 26(5):757-66, 2007.
121. Pyle GG, Paaso B, Anderson BE, Allen DD, Marti T, Li Q, et al. Effect of pretreatment of food gluten with prolyl endopeptidase on gluten-induced malabsorption in celiac sprue. Clin Gastroenterol Hepatol 3(7):687-94, 2005.
122. Daveson AJ, Jones DM, Gaze S, McSorley H, Clouston A, Pascoe A, et al. Effect of hookworm infection on wheat challenge in celiac disease--a randomised double-blinded placebo controlled trial. PLoS One 6(3):e17366, 2011.
123. Crespo Perez L, Castillejo de Villasante G, Cano Ruiz A, Leon F. Non-dietary therapeutic clinical trials in coeliac disease. Eur J Intern Med 23(1):9-14, 2012.
Artículos publicados por el autor
(selección):
Definitions and diagnostic criteria of latent and potential coeliac disease. Common Food Intolerances I: Epidemiology of Coeliac Disease. Karger 2:119-127, 1992
Clinical and pathological spectrum of coeliac disease Gut 34:150-151, 1993
Secretory antibody pattern of coeliac disease: occurrence in patients with normal jejunal biopsy histology Gastroenterology 104:1263-1272, 1993
Gamma/delta T cell receptor expression by intra-epithelial lymphocytes in relation to other indices of potential coeliac disease Gut 35:476-482, 1994
The pattern of cytokine expresión determines the degree of mucosal damage Gut 56(56):441-443, 2007
|
|

