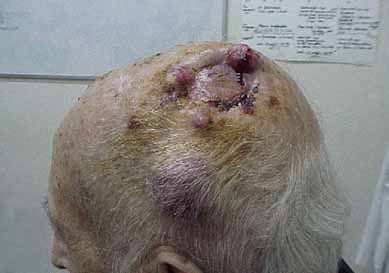
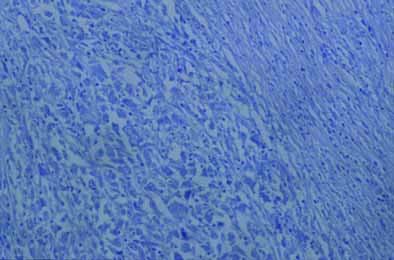
UN DILEMA DIAGNOSTICO EN UN PACIENTE CON LINFOMA DE LENTA EVOLUCION Y LA APARICION DE UN SARCOMA HISTIOCITICO DE GRAN MALIGNIDAD
La sospecha de un sarcoma histiocítico obliga al diagnóstico diferencial con neoplasias de origen linfático, por lo cual resulta esencial la aplicación de técnicas de inmunohistoquímica.
Institución del autor
Hospital Francés, Buenos Aires, Argentina
Coautores
Mariano Adrián Forlino*
Médico Especialista en Medicina Interna, Jefe de Trabajos Prácticos de Farmacología de la Facultad de Medicina de la Universidad de Buenos Aires, Hospital Francés, Buenos Aires, Argentina*
Primera edición en siicsalud
30 de marzo, 2010