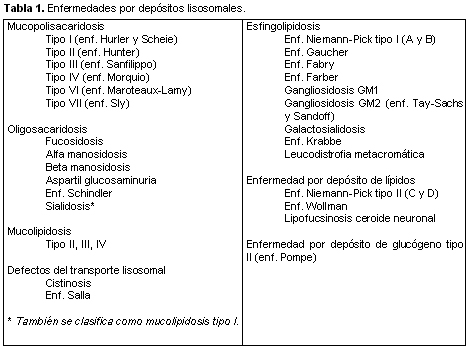
GANGLIOSIDOSIS GM-1 O ENFERMEDAD DE LANDING. PRESENTACION DE UN CASO DE DEBUT NEONATAL
La gangliosidosis GM1 es una esfingolipidosis, de herencia autosómica recesiva. La superposición clínica entre los diferentes trastornos del metabolismo lisosomal hace difícil el diagnóstico diferencial, por lo que es deseable la comunicación y publicación continua de los casos nuevos.
Institución del autor
Hospital Costa del Sol, Marbella, España
Coautores
Alicia Martin Torrecillas* Juan Antonio Ruiz Moreno* Maria Gonzaléz Lopéz* Genoveva Del Rio Camacho*
Pediatra, Hospital Costa del Sol, Marbella, España*
Primera edición en siicsalud
24 de agosto, 2010