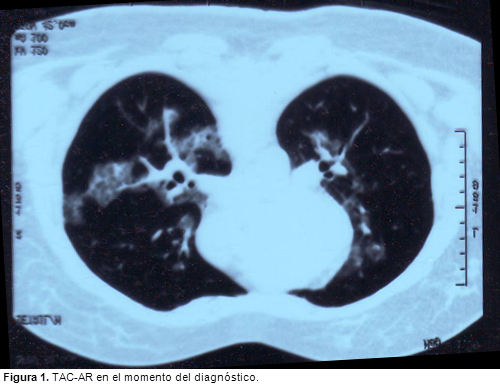
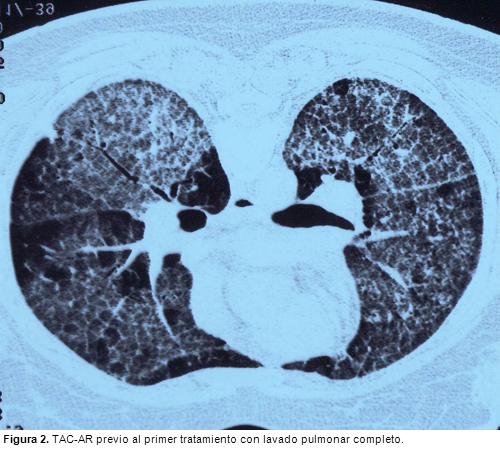
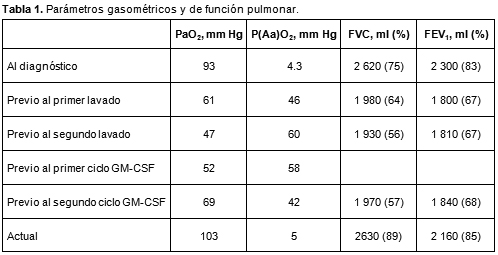
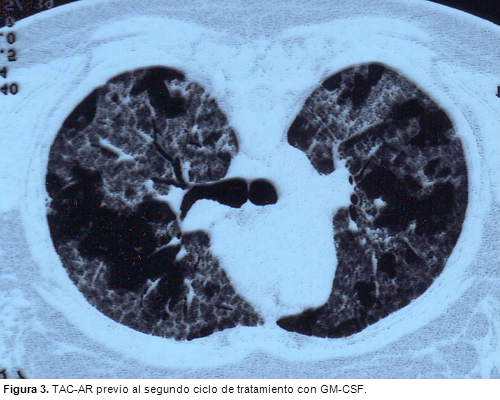
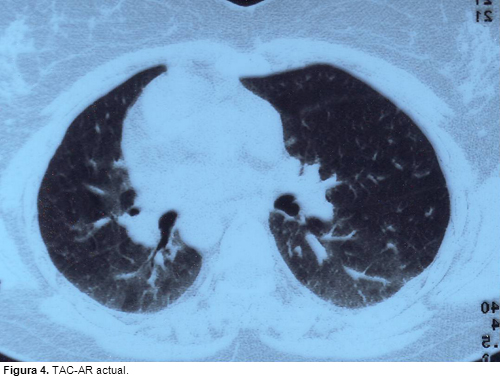
LIPOPROTEINOSIS ALVEOLAR: EFICACIA TRAS EL SEGUNDO CICLO DE TRATAMIENTO CON FACTOR ESTIMULANTE DE COLONIAS DE GRANULOCITO-MACROFAGOS
En el tratamiento de la lipoproteinosis alveolar puede ser eficaz la administración de GM-CSF. En caso de recaídas posteriores a la respuesta inicial, el re-tratamiento con el mismo fármaco constituye una opción útil.
Institución del autor
Hospital Clínico Universitario de Santiago de Compostela, Santiago de Compostela, España
Coautores
Luis Valdés Cuadrado* Antonio Pose Reino** Enrique Temes Montes*** Pedro Alvarez-Calderon Prat**** Sandra Blanco González*****
Doctor en Medicina y Cirugía, Especialista en Neumología, Hospital Clínico Universitario de Santiago de Compostela, Santiago de Compostela, España*
Doctor en Medicina y Cirugía, Especialista en Medicina Interna, Hospital Clínico Universitario de Santiago de Compostela, Santiago de Compostela, España**
Doctor en Medicina y Cirugía, Especialista en Medicina Interna, Hospital de Pontevedra, Pontevedra, España***
Especialista en Neumologia, Hospital Clínico Universitario de Santiago de Compostela, Santiago de Compostela, España****
Doctor en Medicina y Cirugía, Especialista en Cirugía Torácica, Hospital Clínico Universitario de Santiago de Compostela, Santiago de Compostela, España*****
Primera edición en siicsalud
25 de junio, 2010