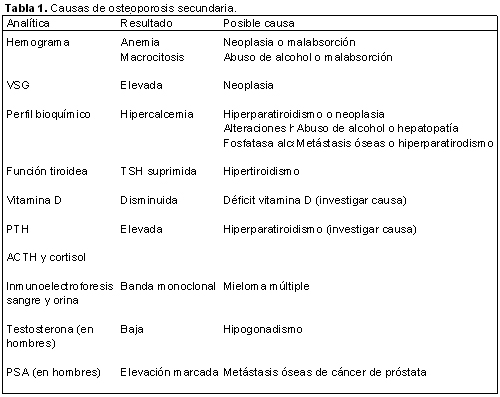
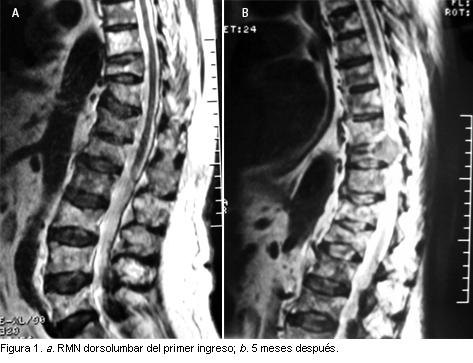
MIELOMA MULTIPLE NO SECRETOR
El mieloma no secretor representa menos del 1% de todos los mielomas. Su peculiaridad reside en la ausencia de componente monoclonal tanto en suero como en orina. Se presenta un caso de la enfermedad de difícil diagnóstico.
Institución del autor
Hospital Clínico Universitario Lozano Blesa, Zaragoza, España
Coautores
Patricia Gracia García* Rocio Zamora González-Mariño** Juan Ignacio Pérez Calvo***
Médico interno residente de psiquiatría, Hospital Clínico Universitario Lozano Blesa, Zaragoza, España*
Médico interno residente de nefrología, Hospital Clínico Universitario Lozano Blesa, Zaragoza, España**
Jefe de sección medicina interna, Profesor asociado médico, Hospital Clínico Universitario Lozano Blesa, Zaragoza, España***
Primera edición en siicsalud
18 de agosto, 2010