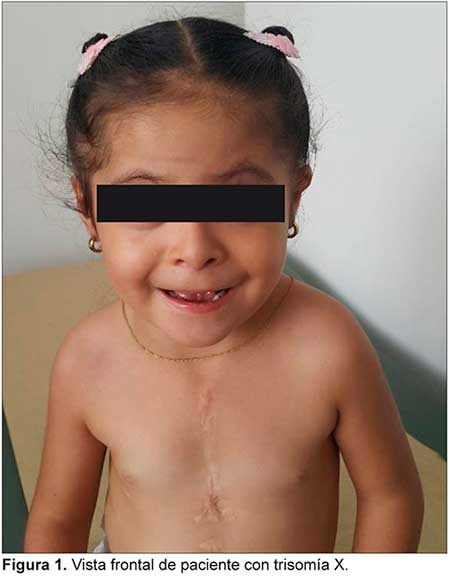
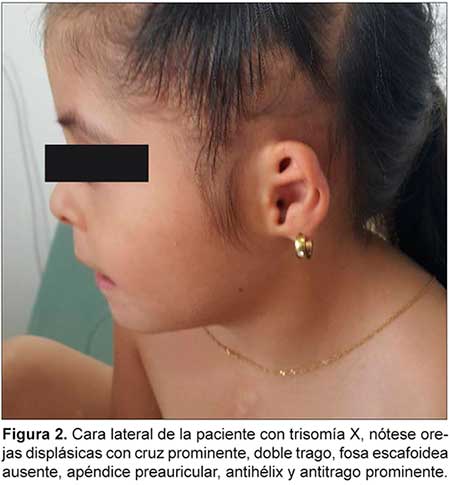
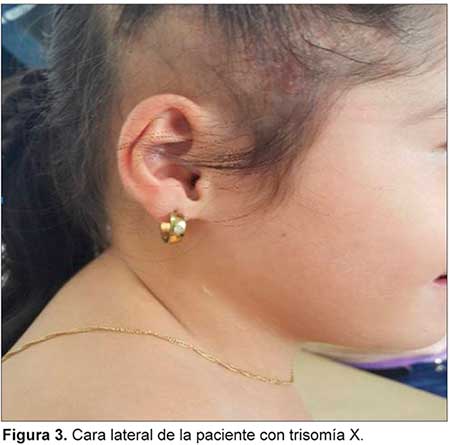
TRISOMIA X ASOCIADA CON CARDIOPATIA COMPLEJA Y MALROTACION INTESTINAL
Se describe un caso de trisomía X asociada a cardiopatía compleja y malrotación intestinal, características descritas por primera vez en un mismo caso hasta el momento. La trisomía X es la aneuploidía más frecuente de los cromosomas sexuales en niñas nacidas vivas, sin embargo, se estima que sólo se diagnostica el 10% de las mismas. Su espectro fenotípico es variable.
Institución del autor
Universidad ICESI, Cali, Colombia
Coautores
Harry Pachajoa*
Universidad ICESI, Cali, Colombia*
Primera edición en siicsalud
20 de enero, 2020