DEFICIENCIA DE COBALAMINA C DE INICIO TEMPRANO: PRESENTACION DE CASO CLINICO
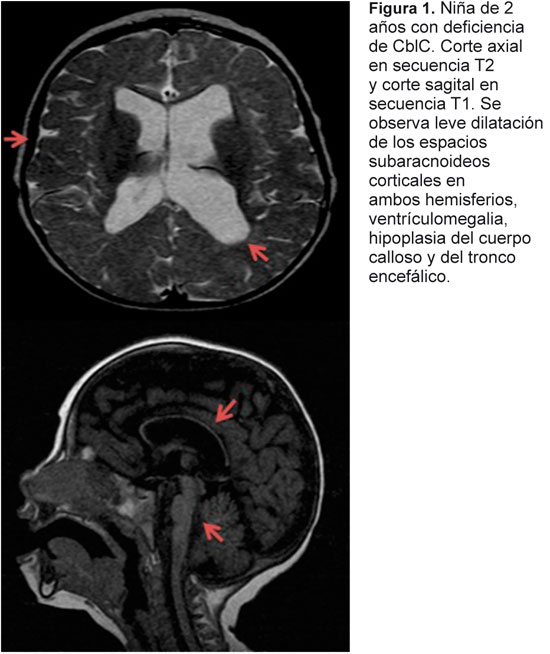
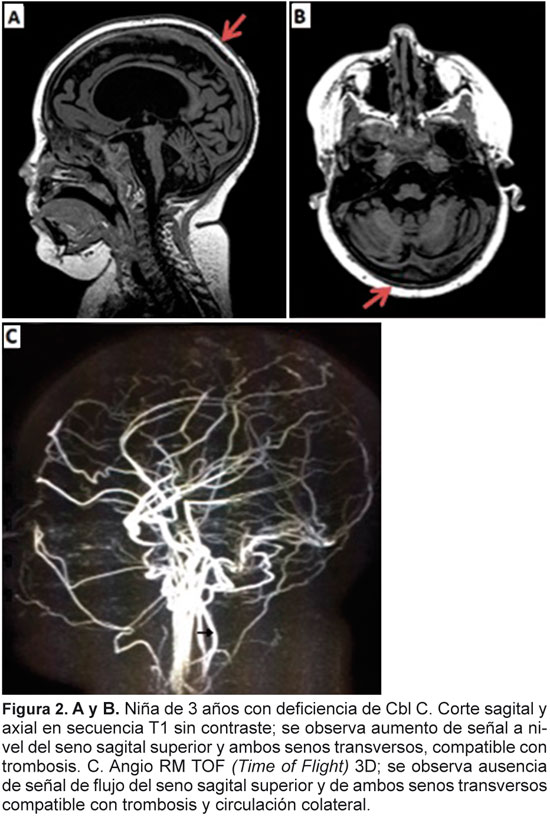
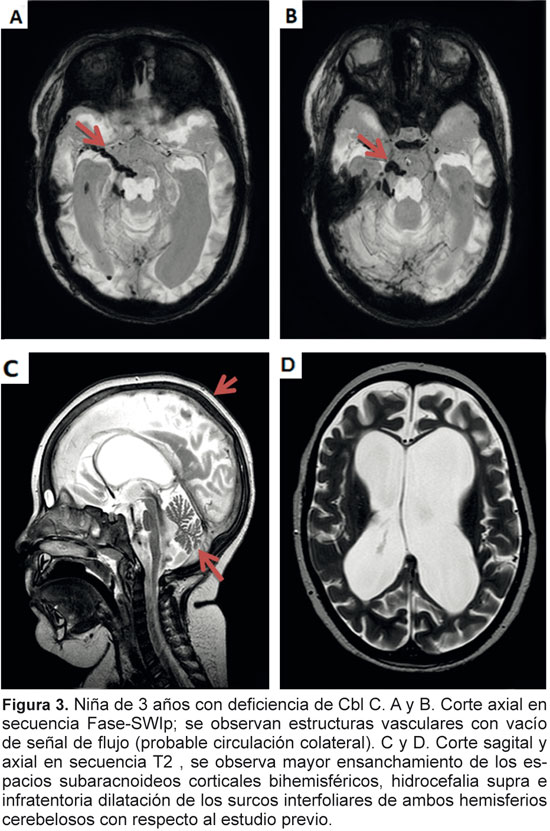
Informamos un caso de deficiencia de cobalamina C en el que el inicio de los síntomas fue agudo, característica que no es frecuente en este tipo de enfermedades. La aparición de trombosis venosa cerebral en el contexto de hiperhomocisteinemia está ampliamente descrito, sin embargo, en este grupo no surge como una complicación frecuente. El abordaje de estas enfermedades debe incluir la sospecha de su existencia por medio de imágenes del sistema nervioso central y de análisis de laboratorio.
Institución del autor
Hospital de Pediatría SAMIC Prof. Dr. Juan P. Garrahan, Ciudad de Buenos Aires, Argentina
Coautores
Hernán Eiroa* Carlos Rugilo** Yeny Blanco***
Médico, Hospital de Pediatría SAMIC Prof. Dr. Juan P. Garrahan, Ciudad de Buenos Aires, Argentina*
Médico Neurólogo, Hospital de Pediatría SAMIC Prof. Dr. Juan P. Garrahan, Ciudad de Buenos Aires, Argentina**
Médica, Hospital de Pediatría SAMIC Prof. Dr. Juan P. Garrahan, Ciudad de Buenos Aires, Argentina***
Primera edición en siicsalud
23 de septiembre, 2019