MUTACION EN EL GEN TMEM70: UNA FORMA DE ACIDURIA 3-METILGLUTACONICA CON FENOTIPO VARIABLE. PRESENTACION DE DOS CASOS CLINICOS
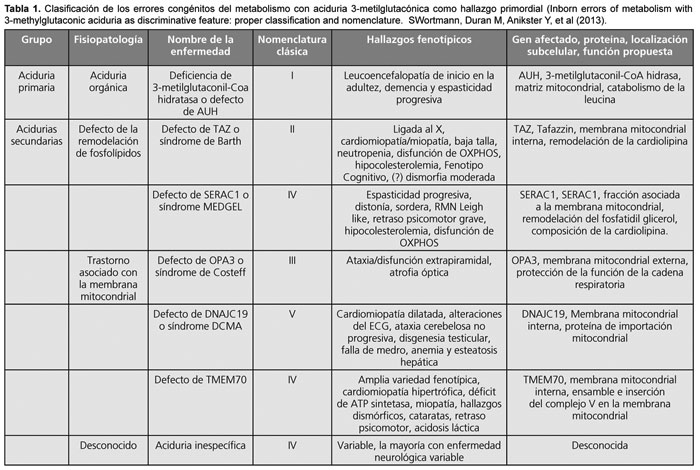
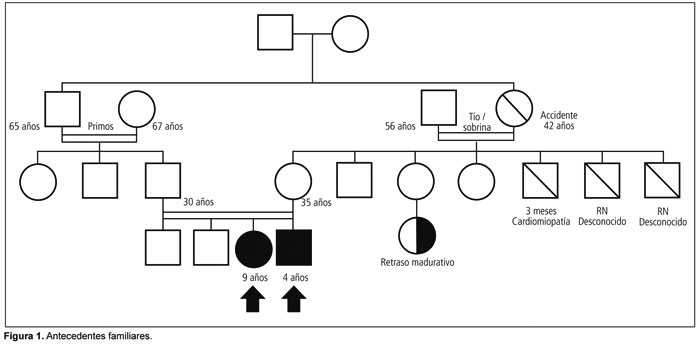
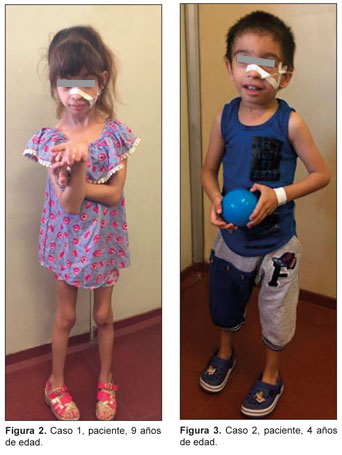
Es nuestra intención aportar la experiencia de dos pacientes con diagnóstico de aciduria 3-metilglutacónica con mutación en el gen TMEM70, seguidos en el Hospital SAMIC Prof. Dr. Juan P. Garrahan con el fin de poder difundir el conocimiento de este tipo de enfermedades, para de esta forma aumentar su sospecha clínica entre la comunidad médica.
Institución del autor
Hospital de Pediatría SAMIC Prof Dr. Juan P. Garrahan, Ciudad de Buenos Aires, Argentina
Coautores
María Cruz Tubio* Ana Clara Bernal** Hernán D Eiroa***
Médica, Hospital de Pediatría SAMIC Prof Dr. Juan P. Garrahan, Ciudad de Buenos Aires, Argentina*
Médica Pediatra, Hospital de Pediatría SAMIC Prof Dr. Juan P. Garrahan, Ciudad de Buenos Aires, Argentina**
Médico Pediatra, Hospital de Pediatría SAMIC Prof Dr. Juan P. Garrahan, Ciudad de Buenos Aires, Argentina***
Primera edición en siicsalud
29 de junio, 2018