NEUROFIBROMATOSIS TIPO 2 MANIFESTADA COMO SCHWANNOMA VESTIBULAR BILATERAL
La neurofibromatosis tipo 2 es un raro trastorno genético autonómico dominante, de alta penetrancia, caracterizado por la presencia de schwannomas vestibulares bilaterales. El diagnóstico suele ser tardío, en ausencia de antecedentes familiares y estigmas cutáneos, y presenta hipoacusia neurosensorial grave asociada con signos de alteración neurológica.
Institución del autor
Hospital de Algeciras, Algeciras, España
Primera edición en siicsalud
21 de marzo, 2017
Introducción
El schwannoma vestibular es un tumor benigno de la vaina neural del nervio vestibular, que puede ser unilateral, esporádico (95%) o bilateral (5%), y es patognomónico de la neurofibromatosis tipo 2.1 Los shwannomas suelen provocar gran morbilidad, asociada con la compresión de los pares craneales adyacentes y el tronco encefálico, llegando incluso a amenazar la vida. Representan el 6% de los tumores intracraneales, y su incidencia se estima en aproximadamente 10 por millón al año.1,2
La neurofibromatosis tipo 2 se ha ligado históricamente a la neurofibromatosis tipo 1 o enfermedad de Von Recklinghausen, pues constituye un trastorno neurocutáneo con herencia autonómica dominante, pero clínica y genéticamente distintos. Así, la neurofibromatosis tipo 1 está producida por una mutación en el cromosoma 17q11.2, que afecta la piel (manchas cutáneas “café con leche”) y el sistema nervioso periférico (múltiples neurofibromas espinales), mientras que la neurofibromatosis tipo 2 es un raro trastorno secundario a la mutación del cromosoma 22, con escasas manifestaciones cutáneas y alto grado de malignidad, en el que predomina la clínica neurológica y cuyo signo cardinal es el schwannoma vestibular bilateral.2
Presentamos el caso de una adolescente, gemela de otra hermana diagnosticada de neurofibromatosis tipo 2, ambas sin clínica otológica llamativa, y describimos los aspectos más relevantes.
Caso clínico
Paciente mujer de 18 años con hipoacusia del oído izquierdo de varios meses de evolución, remitida por el Servicio de Dermatología para valoración y tratamiento de schwannoma vestibular bilateral detectado en resonancia magnética (RM). Como antecedentes familiares tenía una hermana gemela diagnosticada con neurofibromatosis tipo 2 un año antes, con múltiples schwannomas espinales y meningiomas en el tronco cerebral que le provocaron la muerte; como antecedentes personales presenta schwannomas cutáneos (neurofibromas plexiformes) en el cuero cabelludo y en ambos antebrazos.
La exploración otorrinolaringológica y oftalmológica es normal. La audiometría tonal liminar muestra normocusia en el oído derecho e hipoacusia neurosensorial grave del oído izquierdo. Los potenciales evocados auditivos del tronco cerebral (PEATC) son compatibles con una lesión retrococlear bilateral. La RM craneal muestra lesiones en ambos ángulos pontocerebelosos en el componente intracanalicular, de 4 x 3 cm (derecho) y 2 cm de diámetro (izquierdo), con discreto efecto masa sobre la pared derecha del IV ventrículo cerebral (Figuras 1 y 2).
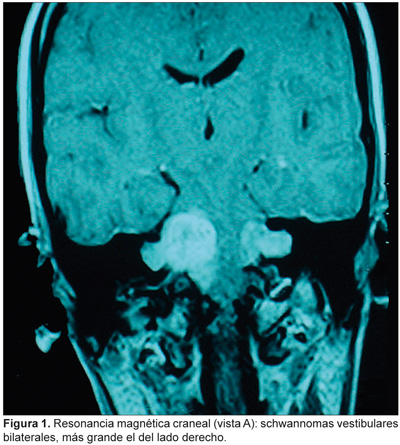
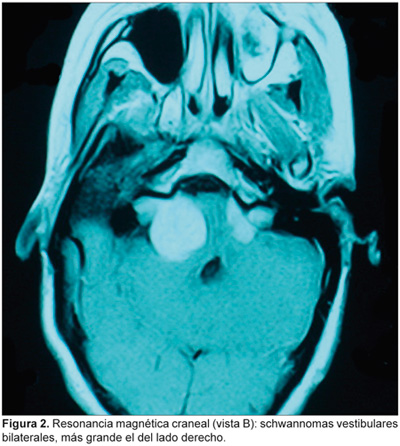
Se le practica craneotomía retrosigmoidea derecha con extirpación subtotal de la lesión, seguida de radiocirugía estereotáxica fraccionada; como secuelas posquirúrgicas quedan parálisis facial y cofosis, ambas del lado derecho.
Discusión
La neurofibromatosis tipo 2 es un trastorno genético autonómico dominante, de alta penetrancia, con un patrón de comportamiento recesivo. Se caracteriza por la presencia de schwannomas vestibulares bilaterales y múltiples meningiomas, aunque pueden asociarse con gliomas, como ependimomas, schwannomas espinales, perineurinoma,3 neurofibromas y cataratas preseniles.
La enfermedad puede ser hereditaria (autosómica dominante) o ser el resultado de una mutación de novo del gen de neurofibromatosis tipo 2 localizado en el cromosoma 22q12.2.2 En estos casos de mutación, tienen mosaicismo (33%), la enfermedad suele ser más grave y clínicamente se localiza en un área del sistema nervioso, apareciendo schwannomas vestibulares unilaterales y meningiomas.4
En 1822 fue descrito por el cirujano escocés James Wishart en un joven panadero, que presentaba sordera bilateral progresiva, que falleció por septicemia tras ser intervenido de un tumor craneal, y en cuyo estudio post mortem se verificó la presencia de un tumor bilateral del VIII par y múltiples tumores de la duramadre.5
No hay predilección étnica. Afecta a 1 entre 35 000 a 40 000 personas; sin embargo, existe una discrepancia entre la incidencia al nacer (1 en 25 000) y la incidencia anual (1 en 2 355 000), debido a que se manifiesta clínicamente hasta la edad adulta o de manera post mortem.6
La presentación clínica es distinta según la edad: los individuos < 15 años tienen schwannomas vestibulares (43%), mientras que en los adultos es más frecuente la mononeuropatía del nervio facial (19 %) y las cataratas (60% al 80 %).7
La característica clínica principal son los schwannomas vestibulares bilaterales (95%), detectados entre la segunda y la tercera década de la vida, que ocasionan hipoacusia neurosensorial progresiva (60%), aunque inicialmente es unilateral. El ritmo de crecimiento es variable, y disminuye con la edad, sin guardar correlación con el tipo de mutación o el grado de gravedad.
El síntoma más frecuente es la hipoacusia asimétrica, de evolución lenta y progresiva, con predominio de tonos agudos sin reclutamiento, acompañado de un deterioro de la discriminación del habla desproporcionada con la pérdida de tonos puros. También experimentan acúfenos, vértigo e inestabilidad; al extenderse comprimen el V y VII par craneal y el tronco encefálico.8 Los schwannomas pueden aparecer en la médula espinal, los nervios periféricos y en otros pares craneales (oftálmico y olfatorio). El 50% de los individuos presentan meningiomas craneales (segundo tumor más frecuente) de mayor ritmo de crecimiento, supratentoriales y múltiples (33%), lo que representa una de las principales causas de morbimortalidad.9 La meningoangiomatosis es rara y asintomática en la neurofibromatosis tipo 2. Las manifestaciones oculares y la afectación cutánea son menos pronunciadas que en la neurofibromatosis tipo 1.10
En ausencia de antecedentes familiares y estigmas cutáneos, el diagnóstico es tardío, con una media de edad de 27 años; se presenta con hipoacusia neurosensorial grave asociada con signos de alteración neurológica, predominantemente trigeminal.
Los criterios diagnósticos actuales incluyen: schwannomas periféricos, tumores espinales, meningiomas craneales y anormalidades oculares, frecuentemente de aparición antes de los schwannomas vestibulares; la mitad de los afectados no tienen antecedentes familiares de neurofibromatosis tipo 2. De esta forma, los criterios diagnósticos serían: schwannomas vestibulares bilaterales; antecedentes familiares de neurofibromatosis tipo 2 más schwannoma vestibular unilateral, o dos de los siguientes: meningioma, glioma, shwannoma, neurofibroma, opacidad lenticular subcapsular posterior juvenil.
Los criterios adicionales están representados por: schwannoma vestibular unilateral más dos de los siguientes: meningioma, glioma, neurofibroma, opacidad lenticular subcapsular posterior; o bien múltiples meningiomas más schwanoma vestibular o alguno de los siguientes: glioma, neurofibroma, cataratas o calcificación cerebral.11
La edad del paciente al comienzo de la clínica es el predictor más importante de enfermedad grave.12
La RM es la prueba diagnóstica principal y de despistaje en familiares. Los potenciales evocados auditivos constituyen una prueba de presunción diagnóstica; sin embargo, la ausencia de umbrales auditivos en la audiometría tonal no debe impedir la práctica de dicho estudio, pues la aparición de una onda I en un enfermo cofótico representaría un signo indirecto de afectación retrococlear. La prueba calórica es la exploración vestibular que más información proporciona, la cual se traduce en arreflexia vestibular bilateral, excepto en el 12% de los sujetos que es normal.13
Debe hacerse diagnóstico diferencial con schwannomatosis (múltiples schwannomas en ausencia de schwannomas vestibulares) y meningiomas múltiples.14
Las distintas estrategias terapéuticas se establecen en función del tamaño y el patrón de crecimiento del tumor, la función auditiva previa y el riesgo de compromiso neurológico. Deben ser tratados en centros especializados por un equipo multidisciplinar experto, y es la piedra angular del tratamiento la conservación o la rehabilitación de la audición y el mantenimiento de la calidad de vida.15
La resección microquirúrgica de los schwannomas debería realizarse lo más precozmente posible para preservar la audición o facilitar, en el futuro, la colocación de un implante coclear o del tronco encefálico.16 Si la audición está preservada y el segundo tumor es < 1.5 cm, se extirparía a los 3 a 6 meses; si no se preserva, se observaría dicho tumor hasta que la audición se pierda o invada el tronco encefálico. La extirpación completa suele ser difícil por su naturaleza quística, la asociación con schwannomas del nervio facial y ser multifocales; este abordaje está indicado en tumores grandes y sintomáticos que provocan compresión del tronco encefálico, hipoacusia progresiva y déficits trigeminal y facial. La lesión accidental del nervio facial puede ser inevitable, a pesar de una monitorización intraoperatoria, debido a la ubicación inexacta del facial en estos tumores.17
Se reserva la radiocirugía estereotáxica cuando la cirugía esté contraindicada y los tumores sean > 3 cm, teniendo en cuenta que el riesgo de malignización secundario a la radioterapia es mayor en la neurofibromatosis tipo 2. El tratamiento conservador se recomienda en tumores con clínica y RM estables.
El implante coclear sería una opción en la hipoacusia profunda y la división coclear del octavo par craneal. Los pacientes < 30 años con schwannoma vestibular unilateral deberían ser controlados por el riesgo de manifestar otros hallazgos de neurofibromatosis tipo 2 (6%), frente al 1% en los sujetos > 30 años.
Además, es fundamental realizar una prueba y aportar consejo genético. Si una mutación no se identifica en el primer grado de una familia, se efectúan audiometrías y RM en los adolescentes; si la mutación está presente, se realiza RM desde los 5 a los 30 años, y después cada 5 años.18
La neurofibromatosis tipo 2 es un raro síndrome que requiere un rastreo sistemático periódico neurológico y oftalmológico, así como una inspección dermatológica desde la infancia y, posteriormente, debe ser supervisado por un equipo multidisciplinar hasta los 40 años, cuando el riesgo de neurofibromatosis tipo 2 en un paciente asintomático es despreciable.




