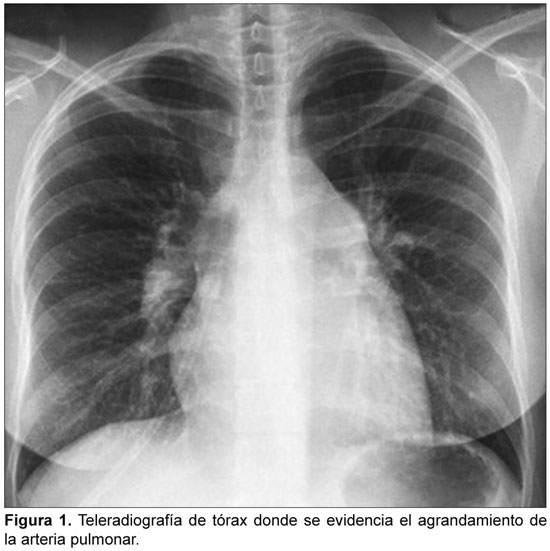
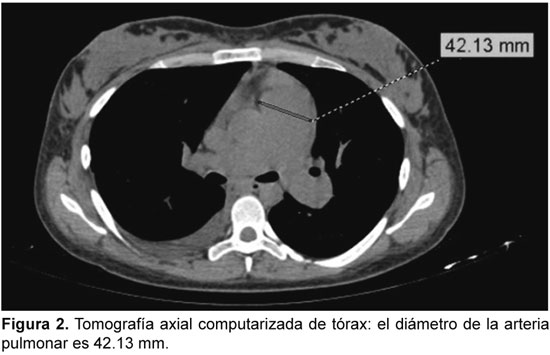
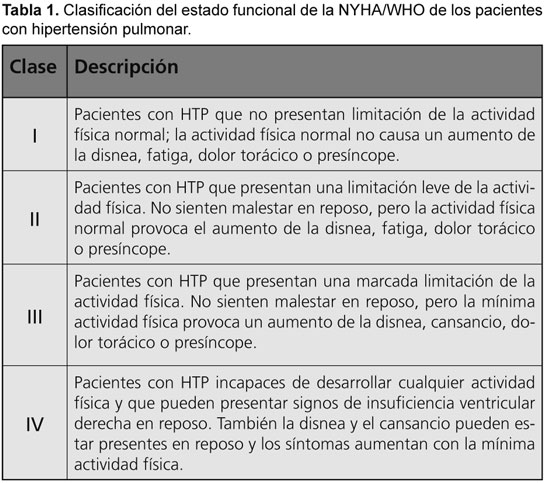
HIPERTENSION PULMONAR ASOCIADA A LUPUS ERITEMATOSO SISTEMICO: REPORTE DE CASO
La asociación de hipertensión arterial pulmonar con lupus eritematoso sistémico es infrecuente. Presentamos el caso de una paciente puérpera con diagnóstico, sin tratamiento convencional, a quien debido a un agravamiento de su enfermedad se le diagnosticó hipertensión pulmonar clase III.
Institución del autor
Clínica Privada Independencia, Munro. Vicente Lopez, Argentina
Coautores
Alvaro Ramirez Toncel* Jhair Martinez Obando*
Medico, Clínica Privada Independencia*
Primera edición en siicsalud
15 de noviembre, 2016