MANIFESTACIONES OTORRINOLARINGOLOGICAS DE LA ESCLEROSIS TUBEROSA
La esclerosis tuberosa es una genodermatosis caracterizada por la formación de múltiples tumores hamartosos benignos, neurofibromas y angiofibromas localizados en la piel, sistema nervioso central, mucosas y otros órganos.
Institución del autor
Hospital Punta de Europa, Cádiz, España
Primera edición en siicsalud
17 de noviembre, 2015
Introducción
La esclerosis tuberosa es una displasia neuroectodérmica, caracterizada por múltiples tumores hamartomatosos benignos: neurofibromas y angiofibromas localizados en piel, mucosas y otros órganos. Se transmite de forma autonómica dominante, con penetrabilidad alta pero incompleta (60% a 70% es esporádica), y expresividad variable. Virchow, en 1860, la describió en el cerebro, en tanto que Vogt, en 1908, definió la triada clásica de epilepsia, angiofibromas faciales y retraso mental. Posteriormente, Von Recklinghausen, Bourneville y Pringle definieron los criterios diagnósticos de la enfermedad.1
La prevalencia varía entre 1 en 6 000 y 1 en 10 000, y su etiología permanece desconocida; se estima que se trata de una mutación en las moléculas de adhesión celular, localizada en los cromosomas 9 y 16, y para otros en el 11 (región 11p23) y el 12. Afecta por igual a ambos sexos y a todas las razas, aunque algunos señalan un ligero predominio entre los varones y en la raza negra.2
La enfermedad expresa gran variabilidad clínica, traducida en múltiples tumores benignos, malformaciones y anomalías que afectan paulatinamente a la mayoría de los órganos, provocando la muerte temprana. Suele asociarse con el síndrome de West (10% al 30%) o encefalopatía mioclónica infantil, caracterizada por la triada clásica de crisis epilépticas clónicas, deterioro o retraso psicomotor y actividad hipsarrítmica en el electroencefalograma.
A continuación, describimos el caso esporádico de esclerosis tuberosa en su forma completa asociado al síndrome de West, y realizamos una revisión de la literatura.
Caso clínico
Paciente mujer de 26 años remitida por Atención Primaria para valorar el crecimiento desmesurado de angiofibromas nasogenianos, que disminuyen progresivamente su visión, desde hace años. Como antecedentes de interés, síndrome de West que apareció a los tres meses; cursa con crisis epilépticas (crisis parciales complejas tónico-axiales) con nódulos subependimarios y túberes corticales cerebrales, retraso psicomotor importante con normalidad de pares craneales y discreta hipotonía global, lenguaje ausente con rasgos autistas sin visceromegalias. No hay antecedentes familiares ni infecciones intraparto ni factores de riesgo al nacer.
A los 10 años se generalizaron las crisis, dejó de andar, y aparecieron múltiples manchas acrómicas (placas seudovitiligoides) en cara, tronco y miembros, y posteriormente angiofibromas faciales, facoma retiniano (provocándole visión borrosa), placa lumbosacra de Chagrin, tumores de Koenen, y angiomiolipoma renal derecho. Actualmente, persisten las crisis generalizadas, y tiene dolores generalizados, por la intensa escoliosis de la columna vertebral.
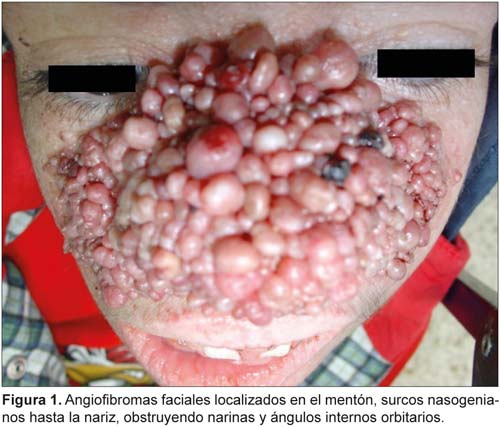
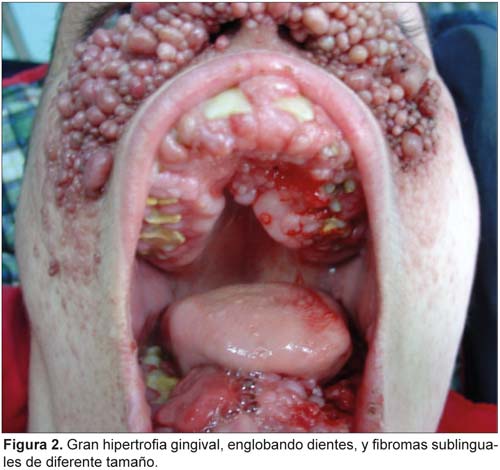
A la exploración otorrinolaringológica se aprecia: angiofibromas faciales (mentón, surcos nasogenianos hasta la nariz, que obstruyen narinas y ángulos internos orbitarios), hemangioma sublingual pedunculado, gran hipertrofia gingival (secundaria a la administración de fenitoína) y fibromas sublinguales (sangrantes al tacto que engloban dientes hipoplásicos), paladar ojival (Figuras 1 y 2). La tomografía computarizada de cráneo-senos paranasales muestra: atrofia cerebelosa, nódulos calcificados subependimarios y subcorticales, displasia fibrosa del peñasco izquierdo y occipucio.
Está en tratamiento sintomático con antiepilépticos, diazepam y analgésicos. Se realiza la exéresis con láser CO2 de los angiofibromas cutáneos faciales.
Discusión
La esclerosis tuberosa es la enfermedad más frecuente asociada con el síndrome de West, son más frecuentes las crisis y el retraso mental en los hombres.3
Clínicamente, aparece en los primeros años de vida, en forma de crisis epilépticas, deterioro psicomotor o ambos, o más tardíamente como crisis comiciales, calcificaciones intracraneales y angiofibromas faciales, y en un tercio es asintomática o paucisintomática.
Las manifestaciones cutáneas tradicionales son las manchas hipocrómicas o acrómicas (85%) en tronco y extremidades y los angiofibromas faciales (adenomas sebáceos de Pringle) son patognomónicos y aparecen en la niñez, entre los 3 y los 10 años, adoptan la forma de mariposa, en los surcos nasolabiales, mejillas, mentón, cuero cabelludo, frente (83% a 90%) y, más raramente, en las orejas; se estabilizan en la adolescencia para persistir toda la vida,4,5 aunque en el caso presentado siguieron creciendo progresivamente.
Las manifestaciones orales (10% a 56%) aparecen a los 4 a 10 años de edad o en la pubertad.6 Las más frecuentes son los fibromas, hiperplasia gingival e hipoplasia del esmalte.7 Los fibromas son nódulos angiofibrosos de color rojo-amarillento o de color normal, de tamaño variable, localizados en las mucosas de labios, mejilla, lengua, paladar y, sobre todo, encías; su incidencia es variable (50% a 69%) con un diámetro medio de 5 mm, y la gravedad de las lesiones dependen de los irritantes locales. Las alteraciones dentales se manifiestan con hipoplasia del esmalte en forma de hoyos o depresiones (100%), paladar hendido, labio fisurado, e hiperostosis alveolar. La esclerosis tuberosa presenta mutaciones de los genes TSC1 y TSC2, que intervienen en la regulación del ciclo celular y procesos neoplásicos, pero no existen estudios que demuestren que existe un riesgo aumentado de cáncer oral8; aunque Fleury (2007) publicó un caso de sarcoma pleomórfico indiferenciado de mandíbula. La hiperplasia gingival se relaciona con antiepilépticos (fenitoína) y la escasa higiene oral.9 Más raramente, se observa tumor odontogénico calcificado, fibroma desmoplásico, hemangiomas mucosos (hemangioma pedunculado en lengua) o intraóseos, mixoma odontogénico, úvula bífida, retraso de la erupción y diastemas.10
El diagnóstico está basado en los criterios diagnósticos de Bourneville-Pringle, seguido de varias pruebas complementarias (recomendaciones de Roach, 1999): tomografía computarizada y resonancia magnética craneal, electroencefalograma, ecografía renal, electrocardiograma, ecocardiograma, examen oftalmológico y dermatológico, pruebas psicomotriz y de desarrollo neurológico, y diagnóstico molecular (identificación de la mutación genética de esclerosis tuberosa).11
El curso y el pronóstico de la enfermedad dependen del tipo de convulsiones y del grado de deterioro intelectual, y en su defecto, de la aparición de insuficiencia renal crónica o de neoplasias malignas.
El tratamiento será el específico de cada una de las manifestaciones clínicas que presente, y deberá ser realizado por un equipo multidisciplinario. Debe completarse con asesoramiento genético.
La esclerosis tuberosa es una displasia neuroectodérmica compuesta por varios tumores habitualmente benignos que requiere ser tratada por un equipo multidisciplinario, enfocado a realizar un asesoramiento genético para aminorar sus manifestaciones clínicas.






