EL SINDROME DE ECTRODACTILIA, DISPLASIA ECTODERMICA Y LABIO/PALADAR HENDIDO. PRESENTACION DE UN CASO Y REVISION DE LA LITERATURA
El síndrome de ectrodactilia, displasia ectodérmica y labio/paladar hendido es un trastorno poco frecuente que se transmite generalmente con un patrón de herencia autosómico dominante.
Institución del autor
Universidad ICESI, Cali, Colombia
Primera edición en siicsalud
5 de diciembre, 2014
Introducción
El síndrome de ectrodactilia, displasia ectodérmica y labio/paladar hendido (EEC, por sus siglas en inglés), es un trastorno poco frecuente que se transmite generalmente con un patrón de herencia autosómico dominante, caracterizado por presentar estas tres manifestaciones cardinales con un grado de expresividad altamente variable y una penetrancia reducida. Fue acuñado como síndrome en 1970 por Rudiger y colaboradores.1 La gran mayoría de los casos son atribuidos a mutaciones en el gen Tp63, aunque se han comunicado casos con otro tipo de alteraciones genéticas.2,3
Se presenta un caso de síndrome EEC que muestra las tres manifestaciones cardinales del síndrome, sin antecedentes familiares, lo cual sugiere presentación esporádica y mutación de novo. Se realiza una revisión de la literatura sobre expresión del síndrome y causas de variabilidad de expresión y penetrancia.
Caso clínico
Paciente de 13 años, hijo de madre con antecedente de cuatro embarazos y cuatro partos (todos varones), sin antecedentes de malformaciones congénitas. No se tienen datos de medidas al nacimiento. Nació con ectrodactilia, labio fisurado bilateral y paladar hendido, para la cual se han practicado cinco cirugías dentro del programa “Fórmula Sonrisa”. La madre refiere retraso en el desarrollo psicomotor con inicio de sostén cefálico a los 8 meses, gateo a los 2 años y marcha a los 3 años. En el momento cursa quinto de primaria y refiere haber perdido tres años.
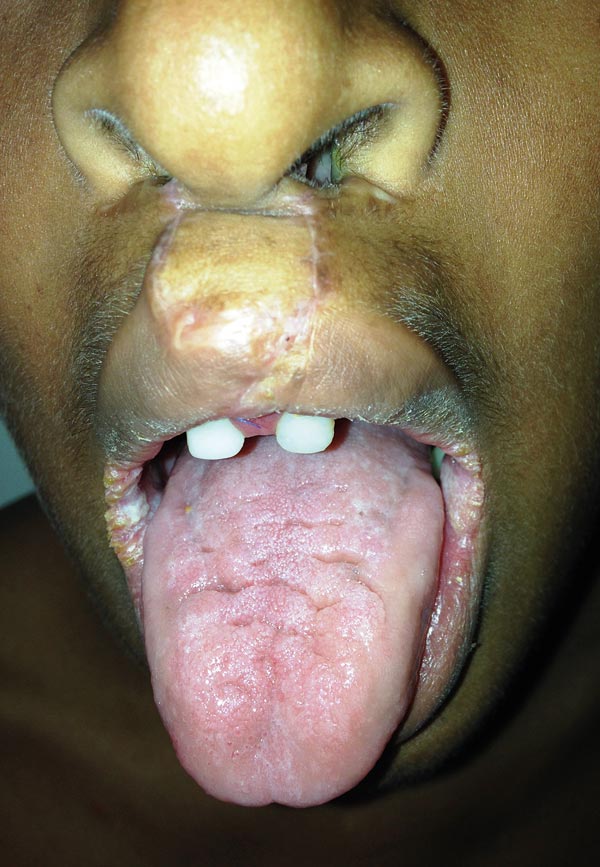
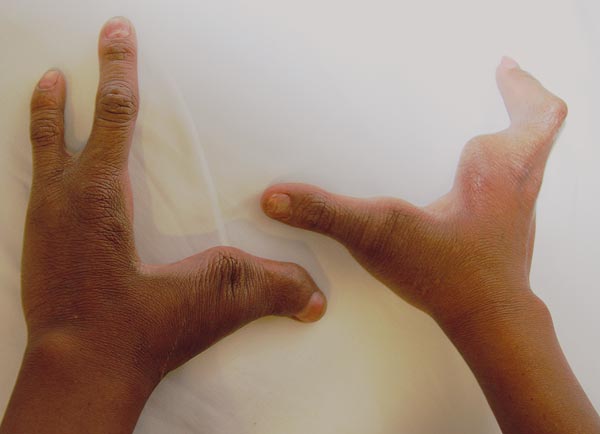
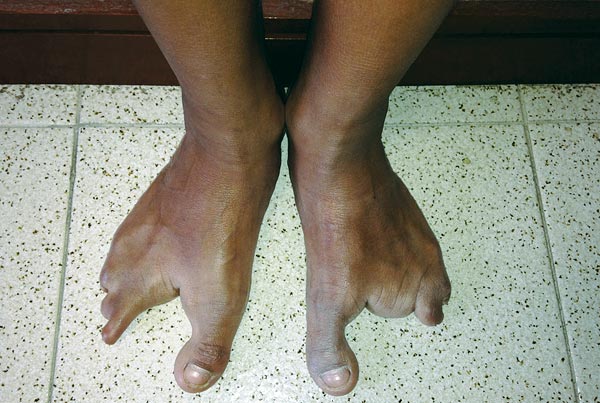
Al examen físico presenta las tres malformaciones cardinales. Cicatriz de labio fisurado bilateral, paladar hendido central y úvula bífida (figura 1); ectrodactilia en manos y pies. Mano derecha con ausencia de segundo y cuarto dedo y sindactilia del tercer y quinto dedo; mano izquierda con ausencia de segundo y cuarto dedo, sindactilia del cuarto y quinto dedo (figura 2). Pie derecho, ausencia del segundo dedo y sindactilia de tercero y cuarto; pie izquierdo, ausencia de segundo y tercer dedo con sindactilia del cuarto y el quinto (figura 3). Piel seca sin dermografismo e hiperqueratosis en comisuras labiales. La madre refiere episodios de otitis y conjuntivitis a repetición. Fisuras palpebrales inclinadas hacia abajo, distancia intercantal interna 3.5 cm, distancia intercantal externa 9.8 cm, distancia interpupilar 6 cm, orejas de 5.5 cm con hélix plegado. A la auscultación, soplo de grado II/IV. Se solicitó ecocardiograma para estudio del soplo, el cual fue normal, y ecografía de abdomen total, normal.
Discusión
El síndrome EEC, es un trastorno raro y poco frecuente con patrón de herencia autosómico dominante, aunque se ha comunicado aparición esporádica en numerosas ocasiones;2,3 fue descrito por primera vez por Cockayne, en 1936, y se acuñó el nombre de síndrome EEC en 1970 por Rudiger. Este cuadro es de penetrancia incompleta, estimada entre 93% y 98%, y expresividad variable.1 Su prevalencia exacta no está determinada pero se estima que se presenta en aproximadamente uno de cada 50 000 recién nacidos vivos.4
El síndrome ha sido clasificado en diferentes subtipos de acuerdo al loci génico afectado; el EEC tipo 1 es el menos frecuente y se debe a mutaciones en el gen EEC1, ubicado en el locus 7q11.2-q21.3; el tipo 3 es causado por mutaciones en el gen Tp63, localizado en el cromosoma 3q27; antes se hablaba también de un tipo 2 asociado al cromosoma 19p12, pero fue descartado por considerarse un modificador fenotípico de la alteración en 3q27.5,6
La mayoría de los casos se han asociado con la mutación del gen p63, éste es un gen de supresión tumoral miembro de la familia del p53, el cual codifica proteínas esenciales para el adecuado desarrollo de las extremidades y de estructuras derivadas del ectodermo, la mutación lleva a un déficit de proteína p63, lo que altera el adecuado desarrollo de éstas. Este mismo gen se ve afectado en otros síndromes asociados con displasia ectodérmica, malformaciones orofaciales y malformaciones de las extremidades; entre éstos se incluyen los síndromes de Hay-Wells, Rapp-Hodgkin, extremidad-mamario, y ADULT. Algunos investigadores consideran estos cuadros como diferentes expresiones de un mismo síndrome, ya que es común la superposición de manifestaciones entre ellos.7,8
Clínicamente, es un síndrome de expresividad variable; los signos cardinales son la ectrodactilia en un 84% de los casos, labio fisurado/paladar hendido en un 68% y displasia ectodérmica en 77% de los pacientes,3 signos que fueron encontrados en este paciente. Recientemente, Vernersson Lindahl y colaboradores demostraron mediante series alélicas de la mutaciones de Trp63 en ratones knock-out, que las hendiduras orofaciales y la displasia ectodérmica se veían favorecidas por pérdida de función de Trp63, mientras que las anomalías de las extremidades eran por ganancia de sus efectos; además informaron que en estos ratones la isoforma TAp63 era un fuerte modificador de los fenotipos en cuanto a penetrancia y variabilidad.9
La ectrodactilia está presente en la gran mayoría de los casos, se caracteriza por la ausencia de uno o más dedos del medio de manos o pies, dando forma de pinzas de langosta. Puede estar asociada o no con sindactilia y, en ocasiones, puede ser sindactilia la única manifestación.10
La presentación de labio fisurado/paladar hendido es variable, pudiendo presentarse el labio fisurado asociado o no al paladar hendido, o únicamente el paladar hendido.11 Se ha determinado que las variaciones en este fenotipo pueden ser atribuibles a la región del gen p63 afectada, de modo que mutaciones en el dominio de unión al ADN se asocian con labio y paladar fisurado, mientras que las mutaciones presentes en la región C-terminal se asocian a sólo labio o paladar fisurado.12
En cuanto a la displasia ectodérmica, su manifestación es también sumamente variable. Es común la obstrucción de los ductos lacrimales, lo que aumenta la susceptibilidad a la aparición de conjuntivitis y otras infecciones.13,14 Puede presentarse xerodermia, hipopigmentación de la piel, zonas de hiperqueratosis, puede asociarse con hipohidrosis y se ha manifestado la presencia de nevus melanociticos en algunos pacientes.3 También son frecuentes las alteraciones del cuero cabelludo, pelo seco y débil, cejas escasas o ausentes y otras alteraciones de los anexos. Algunos síntomas adicionales que pueden presentarse asociados con la displasia ectodérmica incluyen displasia ungueal, odontodisplasia, caries dentales, alteración del esmalte dental, xerostomía, entre otros signos y síntomas que han sido comunicados de manera individual.15
A nivel oftalmológico pueden presentarse otras alteraciones como fotofobia, ulceras corneales, queratitis y blefaritis, presentándose cada vez más problemas con la edad.14 Se han informado en estos pacientes malformaciones de las orejas y alteraciones auditivas, generalmente hipoacusia conductiva, debido a malformaciones del canal auditivo. Aunque en el paciente no se encuentra obstrucción de los ductos lacrimales, sí es claro el antecedente de conjuntivitis a repetición.
Es importante la valoración del tracto genitourinario, ya que las anomalías son frecuentes, incluyen agenesia renal, atresia o estrechez uretral, obstrucción de los uréteres asociada a hidronefrosis, el megauréter es una de las manifestaciones más frecuentes.16 Estas anomalías han sido estimadas entre 30% y 50% de los casos, y pueden estar subestimadas, ya que en muchas ocasiones el estudio se limita a pacientes que refieren sintomatología genitourinaria.17
Algunos autores afirman que las funciones cognitivas no se ven afectadas, aunque pueda haber retraso en el desarrollo del lenguaje, asociado con otras alteraciones como el labio/paladar fisurado o con compromiso auditivo.3 Sin embargo, otros investigadores establecen que hasta un 6.6% de los pacientes pueden presentar déficit cognitivo o retraso mental.15 En este caso, las alteraciones y el retraso en el neurodesarrollo podrían ser atribuibles a las dificultades del lenguaje, por el antecedente de labio/paladar hendido y, por otra parte, como consecuencia de la hipoacusia.
El diagnóstico se basa en la detección de las tres manifestaciones cardinales, con estudios genéticos que lo confirmen. El diagnóstico prenatal puede hacerse mediante ecografía bidimensional o tridimensional del segundo semestre, en especial cuando se presenta un espectro grave en que se encuentre la asociación de la deformidad de las extremidades a paladar hendido. La ecografía 3D es de especial utilidad, ya que permite una mejor visualización de las anormalidades faciales y la ectrodactilia de las manos.18
El manejo de estos pacientes requiere un abordaje multidisciplinario que incluya cirugía plástica y ortopédica, oftalmología, odontología y ortodoncia, dermatología y neuropsicología entre otros.16 Es importante valorar el compromiso genitourinario mediante ecografía, y determinar la presencia de alteraciones auditivas para evitar los trastornos del lenguaje. Debe ofrecerse asesoramiento genético a los pacientes y sus familiares, explicando el patrón de herencia de la enfermedad y el riesgo de tener otros hijos afectados asociados con el mosaicismo germinal.19,20






