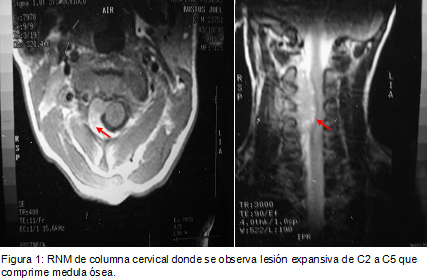
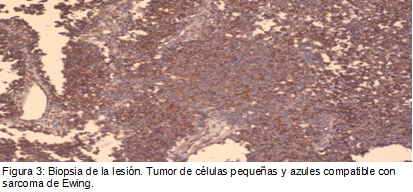
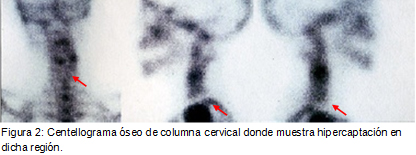
Se presenta el caso de un paciente que comienza con una presentación atípica de sarcoma de Ewing.
Institución del autor
Hospital A. Posadas, Haedo, Argentina
Coautores
Pablo I. Lagrotta*
Médico Pediatra, Jefe de Residentes de Pediatría, Hospital A. Posadas, Haedo, Argentina*
Primera edición en siicsalud
0 de , 0000
(especial para SIIC © Derechos reservados)