SINDROME NEFROTICO FAMILIAR: 12 CASOS
La respuesta terapéutica, independientemente del linaje genético, marca la evolución de la enfermedad.
Institución del autor
Hospital de Niños Dr. Ricardo Gutiérrez, Buenos Aires, Argentina
Primera edición en siicsalud
7 de diciembre, 2009
Introducción
El síndrome nefrótico constituye una causa frecuente de nefropatía infantil. Aproximadamente el 80% de los niños que responden al tratamiento con corticoides por lo general tienen un cuadro histológico denominado de cambios mínimos; el 20% restante corresponde a hialinosis focal y segmentaria (HFS) o a proliferación mesangial difusa, muchas veces resistentes a los tratamientos habituales.1
El cuadro clínico predominante se caracteriza por edemas de diferente magnitud, proteinuria mayor de 40 mg/m2/hora, hipoalbuminemia (menor de 2 g/ml) e hipercolesterolemia. Entre las formas familiares se describe una presentación autosómica dominante (AD) y otra autosómica recesiva (AR) con patrones evolutivos generalmente distintos.2
La forma AR es la más frecuente, afecta con preferencia a niños, tiene diferentes grados de expresión fenotípica y suele progresar rápidamente hacia la insuficiencia renal crónica (IRC).3,4 Por su parte, la forma AD es inicialmente menos grave y su evolución hacia la IRC es más lenta.3 En ambos casos predomina la HFS.
Entre los pacientes con enfermedad autosómica recesiva se han informado también formas corticosensibles, de buen pronóstico y no relacionadas genéticamente con el grupo corticorresistente.
Independientemente de su linaje genético, la respuesta al tratamiento y su caracterización histológica contribuyen a establecer el pronóstico de la enfermedad.5
Pacientes y métodos
Este es un estudio descriptivo de una serie de casos de 12 pacientes (6 pares de hermanos) no consanguíneos con una media de edad de 7 años y 8 meses (2-16 años), 3 mujeres.
Cinco de estos pares de hermanos carecían de antecedentes familiares de nefropatías; el último par presentaba el antecedente materno de insuficiencia renal crónica terminal secundaria a síndrome nefrótico corticorresistente.
Todos los casos se caracterizaron por la presencia de proteinuria mayor de 40 mg/m2/hora, con hipoalbuminemia menor de 2 g/dl e hipercolesterolemia en 2 o más miembros de una familia afectada en 1 o más generaciones.
Consideramos herencia autosómica dominante cuando hay miembros de una misma familia afectados en 2 o más generaciones y autosómica recesiva cuando únicamente son afectados familiares de una generación.3
Todos los pacientes recibieron 48 mg/m2/día de metilprednisona durante 6 semanas y luego 32 mg/m2/día cada 48 horas durante otras 6 semanas. Se consideró remisión el descenso de la proteinuria a 4 mg/m2/hora o menos, con normalización de la albuminemia y del colesterol sérico de acuerdo con los percentilos para sexo y edad. En aquellos que no se logró la remisión se utilizó ciclofosfamida 2 mg/kg/día durante 12 semanas y ciclosporina 150 mg/m2/día.
Se les realizó biopsia renal a 5 pacientes; 4 con síndrome nefrótico corticorresistente (SNCR) y uno con síndrome nefrótico corticodependiente (SNCD).
Clasificamos el daño histológico renal con un puntaje de 0 a 3 acorde con la siguiente escala: ausente, leve, moderada y grave para la esclerosis glomerular, la fibrosis intersticial y la atrofia tubular, considerando el valor mayor o igual a 6 como un índice de riesgo y de potencial menor respuesta al tratamiento.
De los 12 pacientes controlados: 1 par de hermanos evolucionó hacia la insuficiencia renal crónica (IRC), en 3 pares se logró la remisión (1 paciente con ciclofosfamida), en tanto que otro par de hermanos mostraron diferente evolución clínica (1 alcanzó la remisión y el otro evolucionó a IRC); finalmente, los últimos dos hermanos estudiados presentaron SNCD.
La hialinosis focal y segmentaria fue la presentación histológica en 4 de los 5
pacientes sometidos a biopsia y la proliferación mesangial difusa se observó en uno de los 2 últimos hermanos ingresados, a quien se le realizó biopsia.
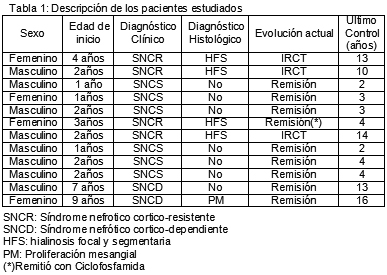
La media del puntaje de daño histológico en los 3 pacientes que evolucionaron hacia la IRC fue de 7, en tanto que en los 9 pacientes restantes fue de 4.
En ninguno de los niños fue posible investigar el linaje genético.
Discusión
En términos generales, la evolución del síndrome nefrótico frente a los tratamientos habituales es variable. Los casos familiares de síndrome nefrótico pueden diferir genética y clínicamente, se ha descrito una forma autosómica recesiva y otra autosómica dominante, con grados diferentes de respuesta terapéutica, de patrón histológico y de evolución clínica.6
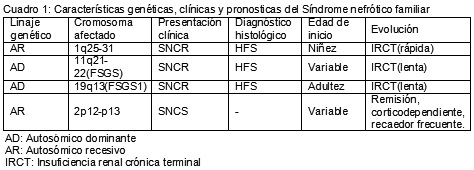
Ciertamente, desde los primeros infomres3 se registran variaciones patológicas en un mismo grupo familiar, con cambios mínimos en algunos pacientes y hialinosis focal y segmentaria en otros de la misma familia.
Por otra parte, ante la aparición del síndrome nefrótico en una misma generación –si bien es poco frecuente– debe siempre pensarse que puede no deberse a factores genéticos sino a la exposición a un mismo factor ambiental.4 Los trastornos del podocito son complejos e involucran factores genéticos y ambientales,7 y a esta altura de las investigaciones parece innegable la existencia de una evolución discordante relacionada con el tiempo y con la respuesta terapéutica, lo cual implicaría una fuerte influencia de mecanismos ambientales y epigenéticos multifactoriales sobre la glomerulosclerosis.
De hecho, publicaciones recientes consideran la presencia de un potencial factor plasmático circulante con acción permeabilizadora sobre la unidad nefronal como el eventual factor ambiental y la mutación ocurrida en el NPHS2 como el factor genético.8
Existe una creciente sospecha de que formas nefróticas que afectan a un mismo grupo familiar pueden tener diferentes respuestas terapéuticas, tal es el caso de la forma recesiva que afecta al gen PLCEI y que causa una forma recesiva del síndrome nefrótico con diversa respuesta al tratamiento inmunosupresor, incluso en hermanos mellizos.
Esta heterogeneidad genética y la gran variabilidad de la enfermedad en individuos de una misma familia demuestran la complejidad genotípica presente, como ejemplo de esto Ghiggeri9 describe 2 pares de mellizos con EFS y distinto curso clínico, lo que resalta la importancia de otros factores aun no identificados plenamente, pero probablemente no genéticos.
Entre las formas AR del síndrome nefrótico corticorresistente se describe un subgrupo de niños con SNCR caracterizados por el inicio temprano de la enfermedad (menores de 6 años) herencia AR y HFS;10 el cromosoma afectado es el 1q25 y la proteína alterada es la podocina. También con linaje AR existen formas corticosensibles no relacionadas con el NPHS2, el mapeo genético identificó la mutación en el cromosoma 2p12-p13.
Por el contrario, en la presentación AD, la HFS es inicialmente menos grave y su evolución hacia la insuficiencia renal crónica es más lenta. Los dos genes alterados son el 19q19 y la proteína codificada es la α-actina 4 (FSGS1), el otro gen alterado es el 11q21-22 (FSGS2); en tanto que la nefrina es codificada en el NPHS1. Las alteraciones de estas tres proteínas mencionadas modifican la unión de los canales iónicos (podocina), la estructura del citoesqueleto (α-actina 4) y la función de los poros de membrana (nefrina).11
Hay también consenso respecto de la existencia de pacientes con HFS y presentación esporádica (mutaciones del gen NPHS2) en cuyos casos el cuadro clínico no permitiría diferenciar las formas esporádicas no familiares de las formas familiares con afección de un solo paciente de una sola generación. Para esta cohorte los factores genéticos son determinantes en la evolución de la podocitopatía no heredada en forma mendeliana, pero el factor ambiental desencadenante (virosis, tóxicos, etc.) es en general necesario.
No obstante el ritmo variable de progresión, la mayoría de los individuos con SNCR evolucionan a IRCT entre la segunda y la tercera década de la vida; por otra parte, se describen formas corticosensibles con evolución favorable de la enfermedad, lo que implica una fuerte correlación intrafamiliar por posible influencia genética.12
De la variabilidad clínica se infiere que las enfermedades asociadas con proteinuria son alélicas; así, en las formas recesivas graves podría haber una pérdida total de la función por mutación de ambos alelos, mientras que una enfermedad moderada tendría lugar por pérdida parcial de la función del mismo gen en uno o ambos alelos. Para ambas formas de transmisión –AR y AD– la penetrancia puede alcanzar el 100% (completa) aunque existen grupos familiares con diferente expresión fenotípica, es decir con grados variables en las manifestaciones clínicas de una misma alteración genética.13
Diez de nuestros 12 pacientes parecen presentar una probable herencia AR si se tiene en cuenta que solamente fue afectada una generación y la desigual evolución en los hermanos podría considerarse como una forma AR de expresión fenotípica variable. Al respecto, Fuchshuber describe grupos familiares corticosensibles, corticodependientes y con recaídas frecuentes dentro de un mismo patrón genético.14
Desde el primer informe hasta la fecha hemos sumado un par de hermanos con síndrome nefrótico corticodependiente y HFS en uno de ellos. En este caso, el hecho de que la madre también estuviera afectada por síndrome nefrótico podría ubicar a esta pareja de pacientes dentro del grupo autosómico dominante; las edades de los dos niños afectados (13 y 16 años) parece confirmar el inicio tardío de esta forma de presentación.
En todos los niños descritos y en aparente concordancia con la evolución del síndrome nefrótico, en general, la caracterización y el grado de daño nefrológico constituyen la marca pronóstica prevalente.
Hasta el momento encontramos un pronóstico relacionado con la respuesta terapéutica y con las características histológicas; una evolución diferente podría corresponder, eventualmente, a una expresión fenotípica variable de una misma alteración genética; no obstante, la identificación de los genes involucrados en la enfermedad tendría importancia fundamental para entender las formas familiares de evolución menos habitual.
El autor no manifiesta conflictos de intereses.






