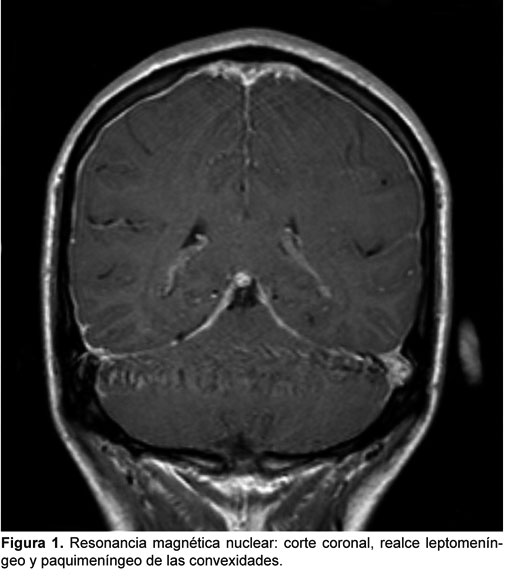
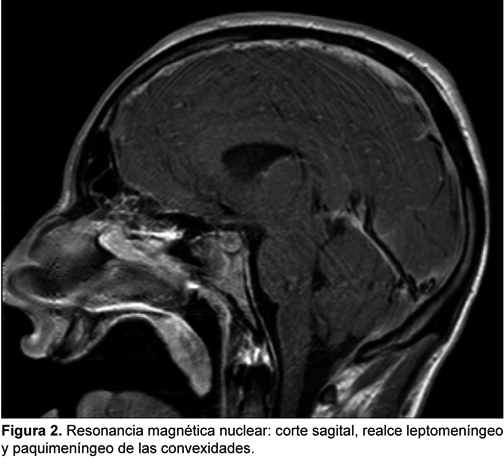
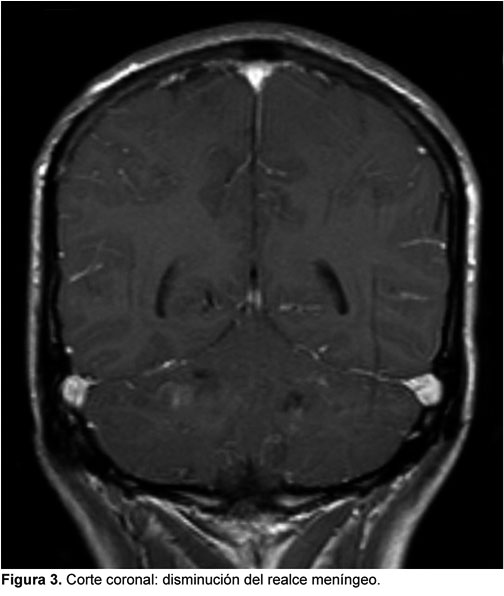
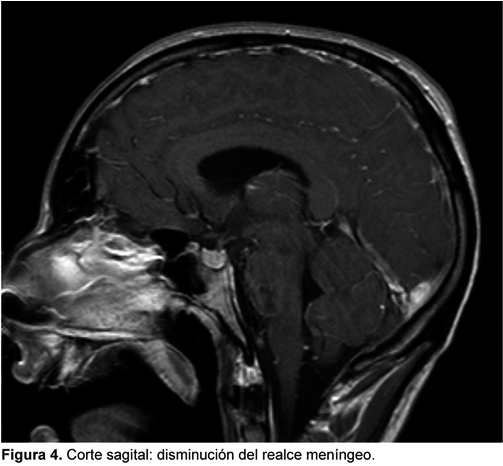
PAQUIMENINGITIS HIPERTROFICA POSINFECCIOSA: PRESENTACION DE CASO CLINICO
La paquimeningitis hipertrófica es una entidad infrecuente que puede ser idiopática o secundaria, y dado que incluso un diagnóstico acertado y un correcto tratamiento pueden dejar secuelas neurológicas graves o incluso producir la muerte, presentamos este caso en el que consideramos que la causa fue una meningitis que no recibió tratamiento adecuado al inicio de los síntomas y que tuvo una respuesta satisfactoria al tratamiento con corticoides inicialmente en altas dosis.
Institución del autor
Clínica Privada Independencia, Munro - Vicente Lopez, Argentina
Coautores
Wilson Manjarrez Coello* José Sabalza Castilla* Fernando Andrés García** Marcela Barón Salgado*** Vidal Tumiri Lezcano* Alvaro Ramirez Toncel* Damián Gonzalo Rutolo*
Médico, Clínica Privada Independencia, Munro - Vicente López, Argentina*
Kinesiólogo, Clínica Privada Independencia, Munro - Vicente López, Argentina**
Médica, Clínica Privada Independencia, Munro - Vicente López, Argentina***
Primera edición en siicsalud
23 de mayo, 2019