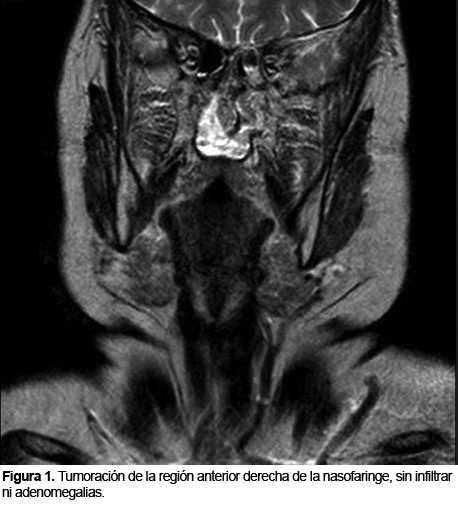
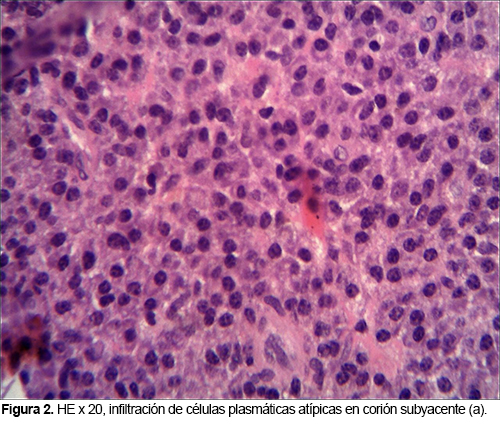
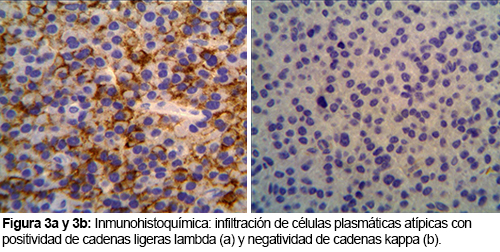
PLASMOCITOMA EXTRAMEDULAR NASOFARINGEO: INFORME DE UN CASO
El plasmocitoma extramedular de nasofaringe es una neoplasia de células plasmáticas inusual, que comprende un 4% de las neoplasias no epiteliales a este nivel. Aparece como una lesión única, de color rojo violáceo, en varones mayores de 40 años y puede ocasionar epistaxis.
Institución del autor
Hospital de Algeciras, Algeciras, España
Coautores
Rosario Guerrero Cauqui* Carmen Benítez García** Regla Gallego Gallegos***
Doctora Especialista en Anatomía Patológica, Hospital de Algeciras*
Doctora Especialista en Anatomía Patológica, Hospital de Algeciras, Algeciras, España**
Doctora, Hospital de Algeciras, Algeciras, España***
Primera edición en siicsalud
1 de diciembre, 2017