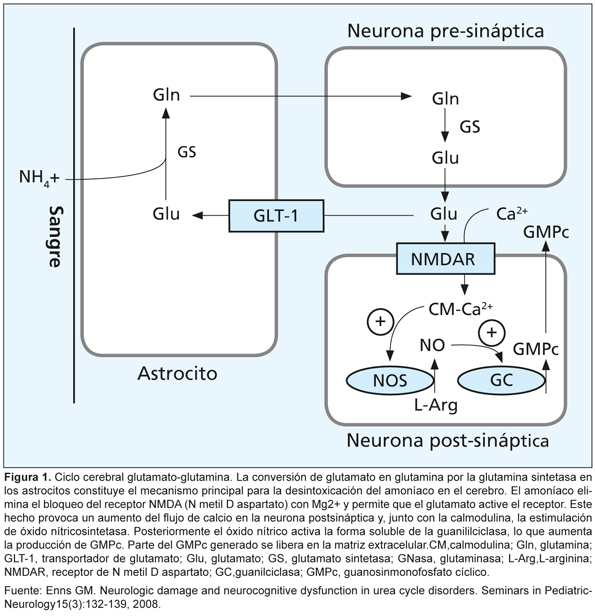
HIPERAMONIEMIA SECUNDARIA A INFECCION URINARIA EN PACIENTE CON MALFORMACION DEL TRACTO URINARIO EN PEDIATRIA: A PROPOSITO DE UN CASO
En general, la hiperamoniemia se asocia con un trastorno del ciclo de la urea; sin embargo, se debe evaluar la técnica de toma y procesamiento de la muestra para hacer un diagnóstico adecuado. Este caso sugiere que los pacientes con vía urinaria dilatada e infectada por un microorganismo productor de ureasa tienen riesgo de presentar hiperamoniemia, situación que puede resolverse con la administración adecuada de antibióticos y la desobstrucción del tracto urinario.
Institución del autor
Hospital de Pediatria S.A.M.I.C. Prof. Dr. Juan P. Garrahan, Ciudad de Buenos Aires, Argentina
Coautores
Hernán Eiroa* Ana Clara Bernal** Verónica Bindi**
Médico, Hospital de Pediatria S.A.M.I.C. Prof. Dr. Juan P. Garrahan, Ciudad de Buenos Aires, Argentina*
Médica, Hospital de Pediatria S.A.M.I.C. Prof. Dr. Juan P. Garrahan, Ciudad de Buenos Aires, Argentina**
Primera edición en siicsalud
24 de octubre, 2017