UN CASO DE NODULOS CUTANEOS MULTIPLES: LINFOMA CUTANEO DE CELULAS T
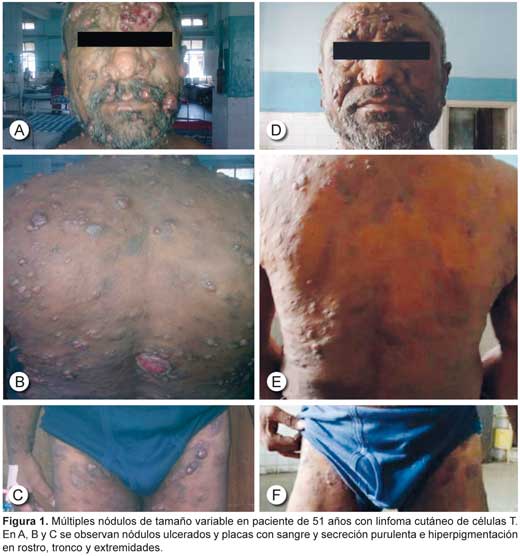
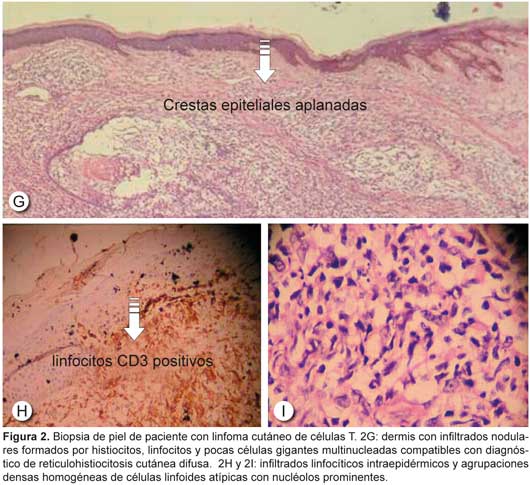
El linfoma cutáneo de células T simula otras formas comunes de trastornos cutáneos, como el eccema, la psoriasis, la dermatofitosis, la sarcoidosis y la neurofibromatosis, cuando la enfermedad está diseminada, de modo que el diagnóstico suele demorarse.
Institución del autor
Byramjii-Jeejeebhoy Government Medical College, Pune, India
Coautores
Rahul S Kulkarni*
MD Medicine, DM Medical Oncology Resident, Gujarat Cancer and Research Institue, Ahmadabad, India*
Primera edición en siicsalud
12 de febrero, 2016