|
INTESTINAL ASPECTS OF CHOLESTEROL GALLSTONE FORMATION
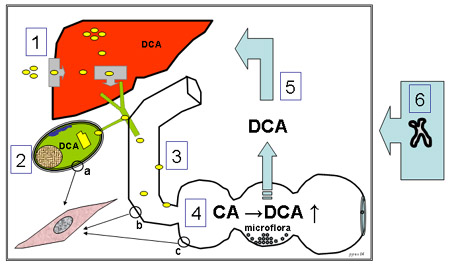
Artículo completo Figura 1. Conceptos actuales acerca de la patogénesis de los litos vesiculares de colesterol (véase el texto para más detalles). 1, hígado: hipersecreción de colesterol (o) y sobresaturación sostenida; 2, vesícula biliar: saturación incrementada de colesterol, incremento en la secreción de mucina, aumento en el desoxicolato, cristalización rápida del colesterol y crecimiento litiásico posterior; 3, vesícula biliar (a), intestino delgado (b), colon (c): incorporación de colesterol dentro del sarcolema, lo que conduce a contractilidad defectuosa del músculo liso; 4, intestino grueso: demora en el tránsito de la bilis e incremento en la transformación de colato en desoxicolato por las bacterias; 5, tracto portal: incremento en la recepción hepática de colesterol y secreción biliar de desoxicolato; 6, un escenario genético puede influir varias de las vías mencionadas más arriba. Leyendas: CA, ácido cólico ; DCA, ácido desoxicólico.El papel del intestino Absorción del colesterol Recientemente se produjeron grandes avances en la comprensión de los mecanismos de la absorción intestinal de colesterol. Durante el proceso digestivo, las sales biliares solubilizan los productos hidrolíticos finales de la digestión de las grasas en el lumen junto con el colesterol proveniente de la dieta y la secreción biliar. Los enterocitos extraen el colesterol de las micelas mixtas enriquecidas en sales biliares únicamente como monómeros.8 Los pasos adicionales incluyen la reesterificación intracelular de colesterol a nivel del retículo endoplasmático de los enterocitos mediante la enzima acylCoA:colesterol aciltransferasa (ACAT) y su transferencia posterior hacia los linfáticos intestinales y probablemente también hacia las partículas HDL en la circulación. El receptor scavenger B (SR-B1) es una proteína de membrana vinculada con el transporte en dirección contraria a través de la HDL.9,10 El SR-B1 también puede encontrarse en los enterocitos intestinales y puede desempeñar un papel como regulador en la absorción intestinal del colesterol.11 Otra proteína intestinal que recientemente fue considerada como reguladora de la absorción es la proteína símil Niemann-Pick C1.12 Otro transportador que se sugirió desempeña un papel en las células intestinales es el ABCA 1. Sin embargo, en ratones ABCA 1 -/- la absorción de colesterol parece reducida sólo en forma leve.10 Un segundo par de transportadores ABC implicado en la regulación de la captación intestinal de colesterol son los semitransportadores ABC G5 y G813-16 descubiertos recientemente. El sistema ABCG5/G8 participa en la transferencia del exceso de colesterol o de otros esteroles desde la célula intestinal nuevamente hacia el lumen intestinal. Las mutaciones en los genes que los codifican causan sitoesterolemia, con absorción incrementada de esteroles vegetales y colesterol. El sistema ABCG5/G8 se halla controlado por el receptor nuclear LXR. Los semitransportadores ABCG5/G8 también están presentes en la membrana canalicular hepatocitaria donde funcionan probablemente como proteínas transportadoras de colesterol.16-18 Se halló que los polimorfismos de ABCG5/G8 afectan la incidencia de la formación de cálculos de colesterol en los seres humanos, ya sea a través de sus efectos sobre su absorción intestinal o por la influencia en la secreción biliar de colesterol.19 Otros factores pueden afectar la absorción intestinal de colesterol20 y el riesgo de enfermedad litiásica. El incremento de la hidrofobicidad del conjunto de sales biliares (más desoxicolato) aumenta la absorción de colesterol en modelos animales.21,22 Los fosfolípidos intestinales también pueden desempeñar un papel. Los ratones carentes de Mdr 2 (-/-) muestran absorción intestinal de colesterol disminuida. En otro modelo animal, en ratones privados de la enzima 7-α colesterol hidroxilasa, tanto el conjunto de sales biliares como la producción biliar de sales biliares se hallan reducidas en forma marcada. La consecuencia de todo esto es que el colesterol se absorbe mínimamente debido a la deficiencia de sales biliares.24 En diferentes estudios recientes en animales se destacó la importancia de la absorción intestinal de colesterol. Los ratones C57L susceptibles de enfermedad litiásica que son expuestos a una dieta litogénica absorben colesterol en forma significativamente mayor (37 ± 5%) que los roedores AKR resistentes a los litos (24 ± 4%).25,26 El tiempo de tránsito intestinal también parece influir en la absorción, es decir, se absorbe menos colesterol con el aumento del tránsito, tanto en seres humanos27 como en modelos animales que incluyen conejillos de Indias hipercolesterolémicos28 y ratones con deficiencias en el receptor para la colecistoquinina A (CCK-A), de acuerdo con lo informado en forma preliminar por Wang y col.20  La motilidad y el incremento del desoxicolato intestinal El conjunto de sales biliares en los seres humanos está formado fundamentalmente por colato y quenodesoxicolato (ambas sales biliarias “primarias” sintetizadas a partir del colesterol en el hígado) y por la sal biliar hidrófoba “secundaria” (desoxicolato) que se forma en la porción distal del intestino delgado y en el colon por la 7-α deshidroxilación bacteriana del colato. El desoxicolato es absorbido en forma parcial desde el lumen intestinal y se une a la circulación enterohepática de sales biliares luego de su conjugación con taurina o glicina en el hígado. El desoxicolato representa del 10% al 30% del conjunto total de sales biliares. Los pacientes con enfermedad litiásica vesicular por colesterol tienen generalmente un conjunto de ácidos biliares ricos en desoxicolato.29-31 El aumento en las cantidades de desoxicolato podría conducir a la hipersecreción biliar de colesterol,32 al incremento en su saturación biliar y a riesgo aumentado para la formación de este tipo de litos.29 En pacientes litiásicos, tanto el tiempo de tránsito del intestino delgado29 como el tiempo de tránsito intestinal global se hallan prolongados en comparación con controles normales. El aumento de los tiempos de tránsito puede intensificar la formación de desoxicolato mediante el incremento de la permanencia intestinal de las sales biliares con aumento en la exposición a la actividad 7- α hidroxilante de las bacterias. Además, el tránsito intestinal prolongado podría intensificar la solubilización de desoxicolato y su absorción a partir del lumen intestinal. De acuerdo con estas hipótesis, el grupo de Dowling34 informó recientemente que los pacientes con litiasis vesicular tienen tiempos de tránsito del intestino grueso más largos, más bacterias anaerobias y grampositivas totales y más actividad de hidroxilación bacteriana en el ciego que los individuos sanos. Además, el grupo de pacientes con enfermedad litiásica mostró valores más elevados de pH dentro del colon proximal, lo que resultó en un aumento en la solubilización del desoxicolato. Todos estos factores pueden promover la formación de desoxicolato y su absorción a partir del lumen intestinal. Existen diversos mecanismos potenciales por los cuales el enriquecimiento del conjunto de sales biliares con desoxicolato puede promover la formación de una bilis litogénica y la formación de cálculos de colesterol. En primer lugar, el desoxicolato podría incrementar la solubilización del colesterol micelar en el lumen intestinal, con aumento de la absorción intestinal de colesterol como resultado de este proceso. Probablemente este efecto sea más relevante en modelos animales con un conjunto hidrofílico endógeno de sales biliares.25,35,36 En contraste, los seres humanos muestran una composición bastante hidrófoba de su conjunto de sales biliares (10% a 30% de desoxicolato). Por lo tanto, su absorción intestinal de colesterol no es, probablemente, muy dependiente de la composición de sales biliares. Nuevos datos aclararon la regulación de la homeostasis de las sales biliares dentro del hepatocito por los “receptores nucleares huérfanos” como el FXR (receptor X del farnesoide), LXR (receptor X hepático) y shp (del inglés small heretodimer partner). Luego de la absorción intestinal, las sales biliares como el desoxicolato son extraídas o tomadas desde la sangre sinusoidal a través de transportadores específicos en la membrana basolateral del hepatocito. Para evitar concentraciones tóxicas y elevadas de sales biliares intracelulares, el FXR activado por la sal biliar induce la disminución de los transportadores basolaterales mediada por shp37 y el incremento de la bomba exportadora de sales biliares: proteína de transporte dependiente del ATP que está comprendida en el transporte de las sales biliares a través de la membrana canalicular hepatocitaria.38 Con mayor relación con este tema, las sales biliares específicamente hidrófobas como el desoxicolato disminuyen eficazmente la transcripción del ARNm de la enzima colesterol 7-α deshidroxilasa, nuevamente a través de la activación mediada por FXR del shp inhibidor.39,40 Resulta claro que estos efectos intracelulares del desoxicolato deberían tener consecuencias importantes para la producción o salida biliar de lípidos y para la saturación de colesterol. El desoxicolato podría también intensificar la secreción biliar de colesterol por un efecto sobre la membrana canalicular hepatocitaria. La fosfatidilcolina y la esfingomielina son los fosfolípidos principales en la lámina externa de la membrana canalicular. Mientras que la primera está contenida en sitios sensibles a los detergentes, se piensa que la segunda está asociada en lugares lateralmente separados que son relativamente resistentes a los detergentes debido a su baja fluidez. Particularmente las sales biliares hidrófobas como el desoxicolato pueden liberar colesterol a partir de dominios de esfingomielina (más probablemente mediante la disminución de la energía de activación requerida para la desorción del esterol a partir de la membrana), permitiendo así su secreción en la bilis.41 Finalmente, el desoxicolato puede ejercer sus efectos litogénicos de forma local en la bilis. Se ha señalado que el desoxicolato facilita la cristatilización de colesterol in vitro y en la bilis intacta. En modelos biliares con sobresaturación de colesterol, el desoxicolato conduce a una expansión considerable de las zonas que contienen cristales en equilibrio43 y a una fuerte intensificación de la cristalización en condiciones de falta de equilibrio.44 De acuerdo con estas consideraciones, en pacientes litiásicos existen firmes correlaciones positivas entre la cantidad de desoxicolato biliar, por un lado, y el índice de saturación de colesterol29,42 o la velocidad de cristalización de colesterol, por el otro. Además, en diversos modelos de formación de cálculos vesiculares, como en pacientes con acromegalia en terapia con octeotride,45 roedores C57L endocriados25,36 o ardillas terrestres35 sometidas a dietas enriquecidas en colesterol, existen cantidades aumentadas de desoxicolato, así como un aumento en los tiempos de tránsito intestinal durante el proceso de formación de litos. Si la prolongación del tiempo del tránsito intestinal es en realidad un factor notable para la patogénesis de la litiasis vesicular, debería ser posible inhibir factores prolitogénicos mediante la aceleración del tránsito intestinal. En efecto, el metronidazol reduce el desoxicolato biliar; el mecanismo involucra la supresión del metabolismo bacteriano del colato. Como consecuencia, la litogenicidad biliar disminuye46. La lactulosa –disacárido no absorbible– acelera el tránsito intestinal,47,48 lo que reduce el tiempo disponible para la absorción.49 Además, la ingestión de lactulosa provoca acidificación colónica, lo cual reduce la solubilidad del desoxicolato y, posiblemente, de la actividad de las bacterias del colon. De igual forma, las fibras de trigo adicionadas a la dieta de pacientes litiásicos reducen la sobresaturación de colesterol y la proporción de desoxicolato en la bilis,50 mientras que evitar comidas con carbohidratos refinados puede reducir el riesgo de cálculos litiásicos de colesterol.51Veysey y col.52 informaron recientemente que en la acromegalia en tratamiento con octeotride, el agente proquinético cisapride fue capaz de acortar la prolongación del tiempo de tránsito intestinal y de disminuir los niveles séricos en ayunas de desoxicolato. Sin embargo, el cisapride no mostró efectos beneficiosos sobre la incapacidad grave del vaciado de la vesícula biliar en estas circunstancias. Mientras todos estos datos aportan indicios indirectos sobre la prolongación del tránsito intestinal como factor prolitogénico existe un sesgo potencial. Los litos vesiculares son muy frecuentes en el mundo occidental y los síntomas atribuidos a ellos, como el dolor en el cuadrante superior derecho del abdomen (hipocondrio) no son específicos de los cálculos.53 De hecho, esos mismos síntomas también podrían ser causados por constipación. En nuestro departamento, generalmente aconsejamos un ensayo con medidas dietéticas o con laxantes para los pacientes litiásicos con tales síntomas. Los síntomas abdominales específicos pueden haber llevado a la detección ecográfica de los cálculos y los médicos clínicos interesados especialmente en la investigación de estos cálculos pueden haber medido el tránsito intestinal y haber incluido a estos pacientes en sus estudios de casos y controles. En un estudio prospectivo reciente54 se evaluó la asociación entre la frecuencia de deposiciones (utilizada como un indicador del tránsito intestinal) en 79 829 mujeres. Entre 1984 y 1996, se documentaron 4 443 casos de incidencia de enfermedad litiásica. Luego del control realizado por edad y factores de riesgo establecidos, los riesgos relativos en el análisis multivariado para mujeres con deposiciones cada tres días o menos fue de 0.97 en comparación con aquellas que presentaron deposiciones diarias y de 1.0 para las que tuvieron más de una deposición por día. No se pudo determinar ninguna tendencia. Por cierto, este estudio puede criticarse por el hecho de que no se midieron los tiempos de tránsito intestinal para documentar si éste era normal o prolongado. Además, la mayoría de los cálculos son asintomáticos y éstos no son tomados en consideración sobre la base de la colecistectomía para la detección de litos. No obstante, estos datos prospectivos obtenidos en un gran número de individuos no parecen indicar que la prolongación del tránsito intestinal desempeñe un papel principal en la patogénesis de los cálculos vesiculares en la mayoría de los pacientes. Otro vínculo posible entre el intestino y la formación de cálculos en la vesícula biliar podría ser la asociación entre la motilidad intestinal y la de la vesícula en el período de ayuno (“interdigestivo”). Aparte del vaciamiento posprandial de la vesícula biliar comentado más arriba, también existe un vaciamiento vesicular considerable (&126;20%-30%) en situación de ayuno asociado con el complejo motor migratorio gastrointestinal (CMM) y con niveles plasmáticos elevados de motilina. El CMM, un patrón de actividad contráctil que en seres humanos el intestino proximal en situación de ayuno presenta en ciclos de dos a tres horas, está caracterizado por tres fases: en la fase I la actividad contráctil está ausente; la fase II presenta actividad irregular, y durante la fase III existen contracciones intensas, coordinadas y regulares. La duración de esta última fase es de sólo unos minutos pero se cree que es particularmente importante para la propulsión de detritos o desechos no digeribles y para la prevención del sobrecrecimiento bacteriano. Aunque la fase III puede iniciarse en todos los niveles del tracto intestinal proximal comienza generalmente en el antro gástrico o en el duodeno proximal. Durante la segunda mitad de la fase II existe una contracción progresiva de la vesícula biliar, con los volúmenes menores de esta última justo antes de la fase III, lo que coincide con los niveles plasmáticos elevados de motilina. La contracción de la vesícula y el incremento en los valores de motilina son especialmente pronunciados si esta fase III comienza en el antro gástrico. En contraste, si se inicia en el duodeno, la contracción de la vesícula y los niveles plasmáticos de motilina son prácticamente insignificantes.55 Se podría asumir fácilmente que en el estado de ayuno (durante la noche) los riesgos de nucleación en cristales y la formación de cálculos vesiculares se encuentran en sus niveles más elevados. El índice de saturación de colesterol se incrementa más durante la noche, ya que la secreción biliar de colesterol se encuentra disminuida en menor proporción que la secreción de sales biliares. Tampoco existe contracción posprandial de la vesícula biliar y, como resultado, la bilis dentro de la vesícula se concentra progresivamente con un incremento en la propensión para la cristalización de colesterol.56 Formulamos la hipótesis de que la alteración en el vaciamiento interdigestivo de la vesícula (asociada posiblemente con un patrón anormal de CMM y liberación de motilina) podría ser importante en la patogénesis de la formación de litos vesiculares. En consecuencia, comparamos el vaciamiento interdigestivo de la vesícula, el patrón del ciclo de CMM y los niveles de motilina en 10 pacientes con enfermedad litiásica y en 10 controles sanos.57 En contraste con estos últimos, los primeros no tuvieron fluctuaciones significativas del volumen de la vesícula biliar durante el ciclo CMM. Además, la duración de este ciclo fue más prolongada en pacientes con patología litiásica que en individuos sanos (158 ± 17 contra 105 ± 10 min, respectivamente; p < 0.05), debido a una fase I más prolongada (40 ± 5 contra 17 ± 3 min. respectivamente; p < 0.05). En contraste con los sujetos normales, las concentraciones de motilina en el grupo litiásico no fueron diferentes en ciclos CMM cuando la fase III se inició en el antro o en el duodeno. Durante los ciclos de fase III iniciados en el duodeno, los niveles de motilina fueron del doble en los pacientes litiásicos que en los controles (p < 0.002). La conclusión de este estudio fue que los pacientes con enfermedad litiásica por colesterol tienen un patrón anormal de liberación de motilina y del CMM. Su vaciamiento vesicular interdigestivo se halla fuertemente reducido y disociado del CMM. Aunque estas alteraciones pueden contribuir a la formación de litos vesiculares, el potencial de sesgo debería excluirse (según lo analizado más arriba con respecto a la relación entre el tránsito intestinal prolongado, desoxicolato y formación de cálculos). Además, las investigaciones futuras deberán dilucidar la relación entre el vaciamiento interdigestivo de la vesícula, el CMM del intestino y la liberación de motilina. La vesícula biliar La vesícula biliar es el sitio en el cual las anomalías intestinales podrían conducir a la formación de cristales, barro biliar y litos. La ingesta de alimento induce un vaciamiento de importancia de este órgano, hasta del 80% del volumen de la vesícula en ayunas, mediante la estimulación de la liberación de la hormona colecistoquinina desde la pared duodenal. La alteración en el vaciamiento posprandial de la vesícula puede alargar el tiempo de permanencia de la bilis en la vesícula, permitiendo de esta forma que exista mayor tiempo para la nucleación de cristales de colesterol a partir de la bilis sobresaturada. Además, en el caso de un vaciamiento normal, los cristales de colesterol nucleados son liberados hacia el duodeno, mientras que si existe un vaciamiento alterado, estos cristales pueden formar conglomerados de cálculos macroscópicos. En pacientes con enfermedad litiásica se detectaron defectos en el llenado y en el vaciamiento de la vesícula.58 También, el volumen de la vesícula en ayunas se halla frecuentemente aumentado,59-61 lo que apunta a un vaciamiento vesicular interdigestivo defectuoso, pobremente integrado con el CMM intestinal.57 Se encuentra motilidad defectiva en la contractilidad del músculo liso,62,63 en la relajación64,65 o en ambas circunstancias en vesículas que contienen cálculos de colesterol, lo que caracteriza un tipo de leiomiopatía vesicular. Se informan alteraciones en el receptor de colecistoquinina A (CCK-AR) y en la transducción de señales;66,67 la explicación más probable es que el exceso del colesterol biliar es absorbido e incorporado dentro del sarcolema de las células musculares lisas, lo que provoca una alteración en la activación del receptor de la proteína G y una reducción en la contractilidad.63,68-71 Los trastornos adicionales en la enfermedad litiásica colesterolémica debidos a exceso en el colesterol biliar y desoxicolato aumentado incluyen diversos tipos de colecistitis crónica,61,72-74 respuestas anormales al estrés oxidativo,75,76 a mediadores inflamatorios77,78 y a efectos perjudiciales de las sales biliares tóxicas.79,80 Conclusiones Debido a su extensión, la complejidad de las funciones y su integración y la sutil interacción con el sistema biliar, el intestino parece desempeñar un papel importante en el desarrollo de la enfermedad litiásica por colesterol. El aumento de la secreción hepática de colesterol en la bilis parece ser el primum movens; antes de que se desarrollen cálculos en la vesícula biliar, sin embargo, pasos patogénicos adicionales incluyen tanto factores locales (proteínas promotoras de la cristalización, alteración de la motilidad posprandial de la vesícula) e intestinales (proteínas transportadoras de colesterol, tránsito intestinal prolongado, incremento de la sal biliar prolitogénica y secundaria desoxicolato). Más aun, una interrupción de la estrecha relación entre la vesícula y la motilidad intestinal en ayunas (interdigestivo), podría constituir un factor adicional, delineándose también un trasfondo de factores genéticos. Más que nunca, se necesita en forma urgente de investigación original para desenmarañar los mecanismos fundamentales que conducen a la formación de enfermedad litiásica por colesterol y para poder delinear mejores enfoques terapéuticos y estrategias de prevención. Los autores no manifiestan conflictos.
|